The Pharma Choke Point
How to Reduce U.S. Dependence on Chinese Pharmaceutical and Biotechnology Supply Chains
Overview
The U.S. pharmaceutical supply chain faces a threat equal to the “rare earths” challenge already posed by China’s exploitation of its dominance of those critical minerals. The authors provide an archetype model that policymakers and researchers can use to anticipate and mitigate a crisis.
 Thomas J. BollykyCFR ExpertBloomberg Chair in Global Health; Senior Fellow for International Economics, Law, and Development; and Director of the Global Health Program
Thomas J. BollykyCFR ExpertBloomberg Chair in Global Health; Senior Fellow for International Economics, Law, and Development; and Director of the Global Health Program Rush DoshiCFR ExpertC.V. Starr Senior Fellow for Asia Studies and Director of the China Strategy Initiative
Rush DoshiCFR ExpertC.V. Starr Senior Fellow for Asia Studies and Director of the China Strategy Initiative Prashant YadavCFR ExpertSenior Fellow for Global Health
Prashant YadavCFR ExpertSenior Fellow for Global Health Olivia KosloffSenior Fellow, American Economic Liberties Project
Olivia KosloffSenior Fellow, American Economic Liberties Project- Research Associate, Global Health, Economics, and Development
This report represents the efforts of a year-long study group convened by CFR’s Global Health Program and the China Strategy Initiative’s China Policy Accelerator.
Executive Summary
U.S. dependence on China for essential medicines is structural—deeper, broader, and more consequential than conventional market analyses suggest. That dependence began with generic medicines and their ingredients but is now growing in biologics manufacturing, first-in-human trials, and synthetic DNA. That dependence is not simply the result of market conditions but rather decades of Chinese state investment.
The United States (and just about every other nation) faces a growing risk that China will deliberately withhold essential pharmaceutical inputs as a tool of economic or political coercion outside of a military conflict, public health emergency, or natural disaster, as China has done with rare-earth critical minerals. Those risks are greatest for the subset of essential medicines and inputs that China exports directly, which include medications to prevent organ transplant rejections, broad-spectrum hospital antibiotics, and a powerful blood thinner. China also dominates upstream inputs for many other critical medicines, such as the antibiotic amoxicillin and the circulatory stimulant norepinephrine, but those inputs are not exported directly to the United States. China would need to restrict supplies or their use in a third country—typically India—to prevent those inputs from being made into active pharmaceutical ingredients (APIs, the chemical compounds in a drug responsible for its effect) or finished dosage forms (FDFs, the final, consumer-ready version of the drug) and exported to the United States. But it has the means, clandestinely if desired, to do so. And if Chinese authorities choose to act, the adverse consequences of cutting off the supply of key starting materials (KSMs), the building blocks that typically feed multiple API and FDF manufacturers, would reverberate throughout multiple pharmaceutical supply chains.
This analysis aims to provide a replicable framework—an archetype model—for policymakers and researchers they can apply to other products that share the same root causes of dependence on China. We identify three archetypes of pharmaceutical supply chain dependence:
Archetype One: Reliance on China for Raw Materials and Upstream Supplies for Generic Drugs
For many essential small-molecule generic medicines—chemically manufactured drugs, simple in structure, easy to replicate, and primarily taken as a pill—the defining vulnerability is a choke point at the raw material or key starting materials stage that makes downstream diversification illusory. Of the U.S. market, China controls the raw or key starting materials for 94 percent of amoxicillin, 74 percent of heparin, and 70 percent of acetaminophen, ensuring that diversification at a later stage provides no meaningful protection. Transshipment and repackaging of APIs and KSMs thwart federal “Buy America” initiatives. Ensuring access to products in this category requires multiple steps. The United States needs to create a strategic reserve of critical medicines and build allied partnerships to manage near-term acute risks. It also needs to undertake a mix of longer-term supply and demand interventions to make domestic or allied production of KSMs, APIs, and other upstream inputs economically viable.
Archetype Two: Erosion of U.S. Biopharmaceutical Manufacturing and Clinical Trial Capacity to China
For innovative biologics—medicines made from living cells—such as monoclonal antibodies, the risk is not a single upstream choke point but competitive displacement across every stage of the value chain: discovery, clinical development, contract manufacturing, and market access. Chinese firms now hold approximately thirty-two of fifty-five late-stage monoclonal antibody programs globally. The Chinese firm WuXi Biologics is engaged in nearly half of U.S. clients’ drug development programs. China biotech licensing reached $137.7 billion in 2025, a tenfold increase from 2021.
Reducing dependence in this archetype requires taking a number of steps: accelerating first-in-human clinical trials in the United States, funding and incentivizing the adoption of advanced biologics manufacturing technologies, building contract research alternatives in allied countries, bolstering the U.S. biomanufacturing workforce, and creating a system to secure artificial intelligence (AI)–ready biodata and digital chemistry, manufacturing, and controls.
Archetype Three: Dependence on China for the R&D Infrastructure for Sensitive Biotechnology
For DNA synthesis—the process for producing DNA from individual nucleotides in the laboratory—the vulnerability is not disruption in the supply of an existing product, but Chinese control over the research-and-development (R&D) infrastructure underlying future pharmaceutical innovation. One of the three firms accounting for 86 percent of the supply of global synthetic DNA is Chinese, and the other two have a significant presence in China. As AI accelerates the design of novel therapeutics, control over synthetic DNA inputs becomes inseparable from control over the trajectory of U.S. biotechnology broadly. As it stands, the offshoring dynamic that hollowed out small-molecule API manufacturing is set to repeat itself in synthetic biology.
Solutions include improving DNA supply chain security, enhancing transparency and disclosing provenance, increasing federal investment in next-generation DNA synthesis technologies, and bolstering allied cooperation on standard-setting and procurement to encourage the adoption of these technologies.
Common Challenges
Cross-cutting themes apply across the three archetypes. Continuity in federal oversight, funding, and dedicated White House–level coordination is essential to overcoming two decades of government inertia. U.S. pharmaceutical supply chain vulnerabilities were visible in trade and regulatory data for years before reaching crisis levels. Monitoring upstream inputs in critical supply chains can help identify and raise the alarm on an imminent shortage before it happens.1 Allied partnerships, such as the Biopharmaceutical Alliance and European Union Critical Medicines Act, are powerful on paper, but without shared inventories, inspection recognition, and purchase commitments, they serve merely as consultative forums, not meaningful guarantees of critical supplies.
A core challenge is incentivizing the commercial adoption of advanced manufacturing technologies. Every industrial policy tool carries risks of unintended consequences—higher costs, reciprocal retaliation, lock-in of obsolete capacity, or accelerated Chinese substitution. These risks need to be anticipated and mitigated.
China has both the tools and demonstrated willingness to weaponize U.S. pharmaceutical dependence: the structural conditions enabling it run through nearly every tier of the pharmaceutical supply. The question is not whether to act, but if the United States will manage to do so before a crisis makes the cost of decades of inaction unavoidable.
Introduction
For decades the United States has allowed its pharmaceutical supply chain to migrate offshore in pursuit of lower business costs, a process accelerated by regulatory inertia and inadequate public investment. The People’s Republic of China (PRC) and India have been the primary beneficiaries. Today, Chinese manufacturers supply most of the world’s key starting materials (KSMs) and active pharmaceutical ingredients (APIs) for critical medicines Americans depend upon. These range from common antibiotics to blood thinners to emergency room drugs such as heparin.
Over the last decade, China has also surged up the innovation ladder to become a major force in the biotechnology sector, displacing the United States as the go-to setting for critical first-in-human (FIH) drug trials and rivaling the United States in developing and producing innovative and promising new medicines. China has pursued a deliberate industrial strategy to achieve this status: it has subsidized manufacturing capacity, undercut competitors on price, and gradually made itself indispensable to the global pharmaceutical system. The result is a structural dependence that now runs through nearly every part of the U.S. medicine supply chain.
This dependence leaves the United States at risk of calibrated coercion by China. Beijing has already demonstrated across rare-earth elements (REEs), critical minerals, and agricultural products that it will weaponize its control of critical supply chain components in response to diplomatic disputes, trade disputes, and geopolitical pressure. The U.S. pharmaceutical supply chain faces a similar risk. “China is the world’s largest exporter of vitamins and antibiotic raw materials,” the Chinese economist Li Daokui stated in a March 2019 general meeting of the Chinese People’s Political Consultative Conference, a top PRC political advisory group. “Once the export is reduced, the medical systems of some developed countries will not work.”2
Beijing does not need to cut off medicine exports entirely to cause serious harm; it can simply restrict licensing, allow quality to erode, and slow shipments through regulatory friction. All those levers are available today, and none requires a formal declaration or a military provocation to deploy.
At times, shortages of the antibiotic amoxicillin, the anti-coagulant heparin, and the circulatory stimulant norepinephrine have already forced American hospitals to ration care, delay surgeries, and substitute less effective treatments. Such examples highlight the vulnerabilities in the supply chain. Few people understand just how this critical vulnerability came to be, how it could be weaponized by China, and what it would take to reverse it.
Scope and Methodology
The U.S. pharmaceutical supply chain faces a threat equal to the “rare-earths” challenge already posed by China’s exploitation of its dominance of those critical minerals. This is due to the structural dependence on China that exists across the entire production of the critical medicines on which Americans rely and the ways in which China could feasibly weaponize that dependence to create economic, strategic, and political leverage. In this report, we
- assess where the dependence on China currently exists or is likely to emerge in the next five to ten years;
- identify the pathways by which China could exploit that dependence and carry out a weaponization program against the United States;
- outline the strategic, public health, and geopolitical risks of dependence; and
- develop a menu of targeted industrial policy options for mitigating and reducing U.S. dependencies in this sector.
In accomplishing these tasks, this report breaks new ground in several ways.
First, this report focuses on “peacetime weaponization”: the deliberate withholding of pharmaceutical inputs as a tool of economic or political coercion outside of a military conflict, a public health emergency, or other kinetic events. Previous analyses have concentrated on how China might weaponize its dominant market share in aspects of the U.S. pharmaceutical supply chain during a pandemic or a military attack.3 China’s coercive episodes, however, have followed a different pattern: they are triggered by political grievances amid ongoing global strategic competition, target civilian industries with no direct involvement in the dispute, are implemented through informal means to maintain deniability, and escalate over time.4 This report focuses on the risks of this type of weaponization, which may harm access to different critical medicines and require different policy solutions than those available in a military or public health crisis.
Second, this report’s findings, wherever possible, are grounded in original data analysis. To assess the scope of dependence on China for critical medicines, we examined U.S. import trade data by looking at harmonized tariff schedule codes and United States Pharmacopeia (USP) ingredient sourcing records, and mapped the supply chain tier by tier, from raw feedstocks and KSMs through APIs to finished dosage form (FDF) pharmaceutical products. Where transshipment has obscured the true origin of inputs, bilateral trade comparisons have been used to expose the gap between the nominal country of origin and the actual one.
Third, this report focuses on case studies, representing the distinct archetypes of dependence on China that exist in critical U.S. pharmaceutical supply chains. Overcoming the origins of U.S. pharmaceutical dependence on China requires understanding why some essential medicines and ingredients are no longer produced in the United States and why that same shift could happen to others. Rather than attempting an exhaustive audit of every vulnerable medicine and the entire U.S. pharmaceutical landscape, the report aims to create a replicable analytical framework—the archetype model—for policymakers, agencies, and researchers to apply to additional product classes. The three structural archetypes identified, explained in detail below, are each represented by case studies, chosen for their presence on critical medicines lists, documented history of shortages, and analytical usefulness across product classes. The six case studies are: amoxicillin, heparin, norepinephrine, acetaminophen, monoclonal antibodies, and synthetic DNA.
Fourth, this report provides granular, realistic policy options for addressing the root causes of these problems. These policy options are intended to be bipartisan in nature and politically feasible. These recommendations and our underlying analysis benefited immensely from the inputs and insights of a bipartisan CFR study group, with a cross-disciplinary scope that spans China policy, biosecurity, manufacturing, regulation, supply chains, trade, and industrial policy. The group met eight times between June 2025 and May 2026, and its members are listed in the appendix of this report.
A Brief History of China Weaponizing Supply Chain Concentration
Over the last fifteen years, Beijing has purposefully used supply chain concentration as an instrument of statecraft and as a dimension of its national security strategy. As noted in China’s signature Made in China 2025 document, “building an internationally competitive manufacturing industry is the only way China can enhance its comprehensive national strength, ensure national security, and build itself into a world power.”5 China has deployed the centralization and concentration of supply chains as a coercive tool in response to diplomatic disputes, market access demands, and Taiwan-related scenarios, and wielded its powers with enough deniability that attribution is often contested long after the damage is done.
China has deployed the centralization and concentration of supply chains as a coercive tool in response to diplomatic disputes, market access demands, and Taiwan-related scenarios, and wielded its powers with enough deniability that attribution is often contested long after the damage is done.
China pioneered its supply chain playbook in rare-earth elements. Beijing controls roughly 70 percent of global extraction and 90 percent of global processing of these materials. The focus on critical minerals may have entered the zeitgeist fairly recently in Washington, but Beijing began leveraging its market dominance as early as 2010. That fall, following a Chinese fishing boat’s collision with two Japanese vessels near the Senkaku/Diaoyu Islands, Beijing halted REE shipments to Japan without formal announcement, allowing the embargo to operate quietly while creating plausible deniability.6
This pattern of unofficial, deniable restriction is a recurring feature of China’s supply chain statecraft. Rather than issue formal export bans, Beijing slows shipments, delays licenses, and multiplies inspections. This pattern of behavior allows the PRC to benefit from the coercive effect of an export ban while avoiding the potential legal consequences of a declared embargo. More recently, in April 2025, Beijing imposed formal controls on seven heavy REEs and radically curtailed licensing approvals, triggering sharp price surges, factory closures across Western allied nations, and the temporary shutdown of a Ford plant in Chicago.7 Those examples offer a preview of the cascading industrial disruption that pharmaceutical supply chain coercion could replicate at an even more consequential scale.
This model of supply chain weaponization has been extended to other industrial inputs and trading partners. China imposed import restrictions on Australian goods after Canberra called for an independent investigation into COVID-19’s origins. Beijing embargoed Lithuania after Vilnius permitted a Taiwanese representative office to open. At least fifteen countries have experienced a form of Chinese supply chain coercion, a pattern that reflects not isolated episodes but a consistent strategy.8
U.S. dependence on China for critical pharmaceutical inputs is mirrored across many low- and middle-income countries, and it is increasing. As part of the Belt and Road Initiative (BRI), China is financing pharmaceutical manufacturing abroad while providing the technical expertise needed to operate it. By embedding Chinese-origin APIs, key starting materials, equipment, financing, and technical standards into pharmaceutical manufacturing across developing countries, the BRI extends China’s sway globally and creates third-country leverage channels even where direct exports to the United States are limited. In a future crisis, such as a major disease outbreak in Africa, countries with nominal local production capacity could be constrained by losing critical Chinese-controlled inputs. That could compromise that outbreak response, threatening global health security and U.S. interests. This strategy, which provides China with a powerful political and economic tool, extends beyond pharmaceuticals into adjacent sectors such as genomic sequencing and biomedical research infrastructure, where Chinese firms and state-backed initiatives are expanding their global footprint as well.9
Any tier of a supply chain in which China has achieved concentration serves as a potential point of leverage which can be exercised formally or informally, announced or deniable, in response to geopolitical flash points or market access disputes. The pharmaceutical supply chain is not exempt from this dynamic. It is, increasingly, its next frontier.
Pathways for Weaponization and Impact Propagation in the Supply Chain
An Anatomy of Medicine and Its Production
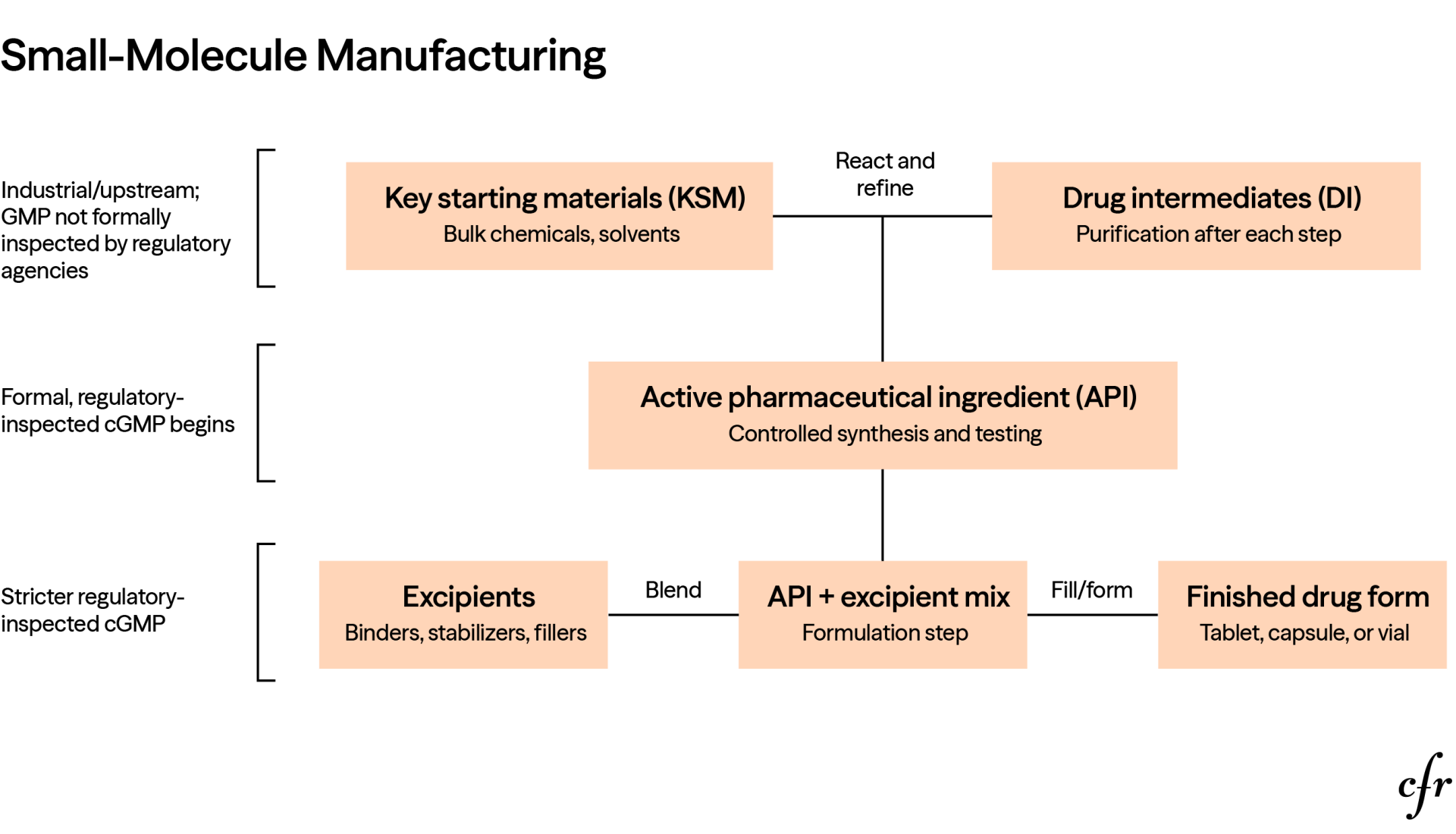
Medicines are produced through a multistep process that begins with basic chemical inputs (key starting materials) that undergo transformation into drug intermediates and ultimately into active pharmaceutical ingredients.
For small-molecule drugs, the production is usually a chemical synthesis pathway, involving stepwise chemical synthesis and purification after each step. The upstream production of KSMs and drug intermediates often looks more like industrial chemical-plant manufacturing. It can involve bulk chemical reactions, solvents, reagents, commodity inputs, and large-scale purification before the material enters the more tightly controlled pharmaceutical supply chain. Regulatory standards become progressively stricter as the process moves downstream, and production stages from APIs to FDFs are under strict good manufacturing practice (GMP). APIs are combined with excipients to create the finished product. Excipients are ingredients such as colorings, emulsifiers, flavorings, lubricants, preservatives, solvents, binders, stabilizers, and release-control agents. Excipients do not have a role in the intended therapeutic effect of the drug, but they are essential for manufacturability, stability, delivery, appearance, and patient usability. After mixing with the excipients, APIs are finished in the form of a tablet or capsule or filled in a vial.
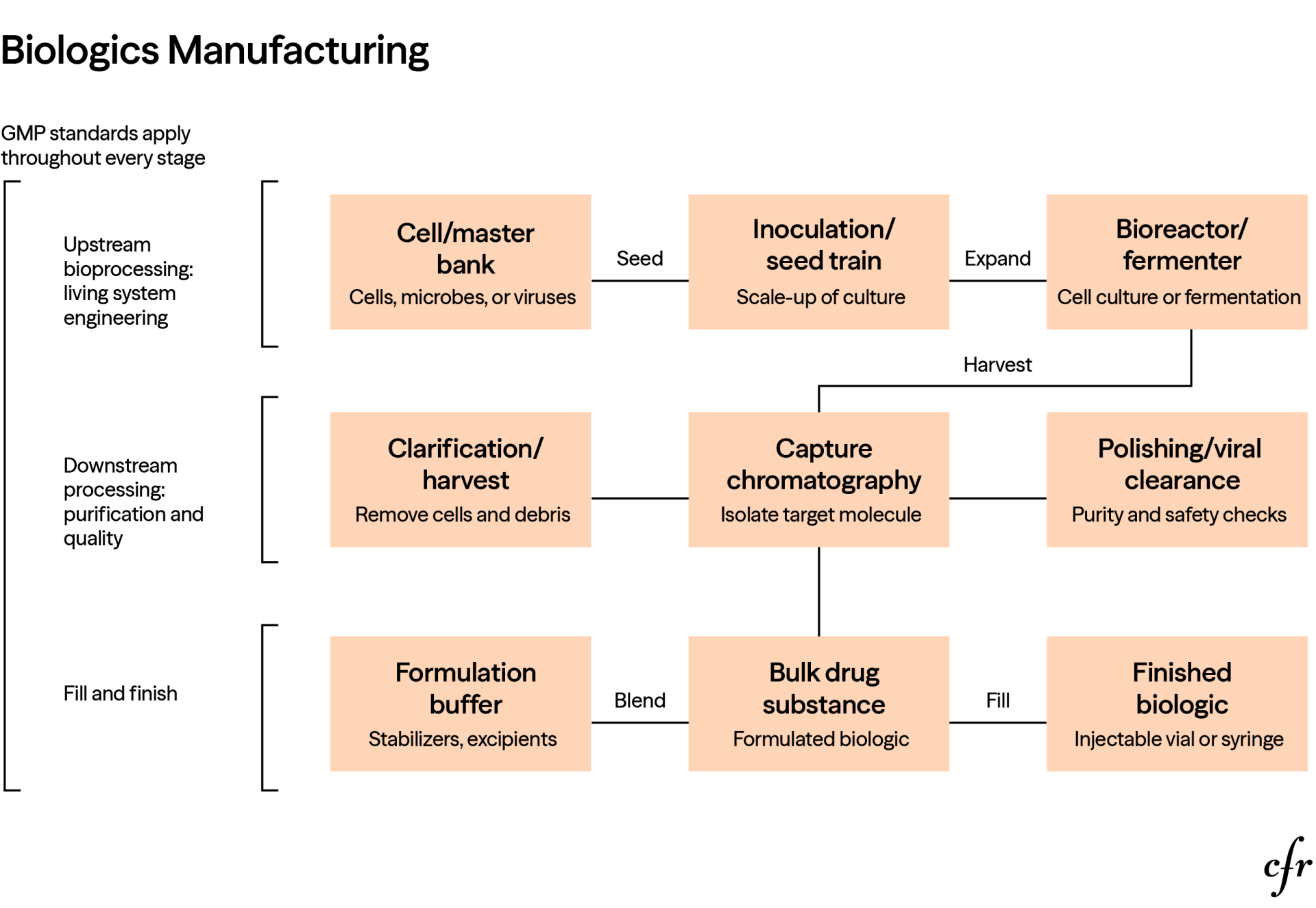
In contrast to small molecules, biologics are made from or with living systems such as microorganisms, cells, proteins, sugars, nucleic acids, tissues, or complex combinations of these. Their manufacturing relies more on cell culture, fermentation, expression systems, and purification than on traditional stepwise chemical synthesis. This sequence requires more tightly controlled processes and quality checks.
The Origins of U.S. Dependence on China for Pharmaceutical Supplies10
U.S. pharmaceutical manufacturing emerged from the dye and chemical industry in the 1930s and expanded dramatically with the increased production of antibiotics in World War II. Because drug and vaccine manufacturing is capital intensive, and the quality, safety, and efficacy of medicines are regulated, it was initially less susceptible to the low-cost locales that lured the garment, consumer electronics, and other labor-intensive industries offshore. Well into the late 1990s, the United States, Europe, and Japan dominated the global market for medicines and their key ingredients.
Change began with the widespread production of low-cost generic medicines—off-patent and unbranded—in the United States. The Drug Price Competition and Patent Term Restoration Act of 1984 established the generic drug industry in the United States by making it easier for pharmaceutical manufacturers to receive approval for a generic drug without having to redo all the expensive clinical trials that supported the original drug’s approval.11 Once a generic drug is deemed bioequivalent to a patented drug listed in the Orange Book, run by the U.S. Food and Drug Administration (FDA), it becomes officially substitutable.12 Because generics are often cheaper than patented medications, many state legislatures have created rules allowing or mandating substitution by pharmacies. Pharmaceutical benefit managers, Medicaid programs, and insurers have done the same, encouraging patients to use the cheapest available FDA-approved generic version by implementing strong financial incentives.13
Consolidation, particularly among pharmaceutical wholesalers, has spurred ruthless generic price competition, in which wholesalers solicit daily tenders and choose the lowest price on offer.14 The wholesalers then compete for customers—hospitals, health systems, retail pharmaceutical chains, and other providers—on price rather than quality, availability, or manufacturing resiliency.15
This combination of a business opportunity to sell ever-cheaper versions of generic drugs and more conducive U.S. laws and policies opened the door to cheap imports from abroad. Shifts in trade policy kicked that door open wider, with the United States entering into a 1994 World Trade Organization agreement exempting pharmaceuticals from tariffs and normalizing trade with China in 2000.16 Environmental concerns played a role as well in shifting manufacturing abroad for the ingredients in many common antibiotics and everyday cardiovascular drugs such as statins. Public alarm over the discovery of hormones in pharmaceutical manufacturing runoff feminizing fish in streams, as well as concern over high concentrations of antibiotics in riverbeds and sewage, prompted new U.S. and European environmental risk assessments and regulations.17 API and KSM production often involves chemical reactions with toxic byproducts, which began to get greater scrutiny from Western regulators and media.
China offered more lax environmental laws, discounted water and power inputs, and, as more API and KSM manufacturing became sourced in that nation, enormous economies of scale. Large plants for manufacturing antibiotics and other pharmaceuticals and their ingredients emerged in China, supported by well-integrated material supply chains that reduced material, shipping, and transaction costs to go along with already low labor-cost advantages. In certain cases, the country pursued predatory industrial policy measures that drove U.S.-based manufacturers out of business, such as when China flooded the global market with low-priced penicillin and vitamin C in the early 2000s.18
In 2021, China’s antibiotic exports constituted one-fifth of global antibiotic exports and nearly half of global antibiotic ingredients exports. The country’s production capacity reached fourteen thousand tons—nearly three times that of India—for amoxicillin, a broad-spectrum penicillin antibiotic used to treat several bacterial infections, including ear, throat, sinus, respiratory, and urinary tract infections, especially in children. In the United States, physicians write more than sixty million amoxicillin prescriptions a year. Although the medicine is sourced from generic manufacturers in Europe, India, Israel, Jordan, and a small Tennessee facility, most of them rely on APIs made in China, which are primarily produced in Jiangsu, Shanghai, and Zhejiang. If China withheld amoxicillin API supplies for geopolitical reasons, as it has with rare-earth elements, or a natural disaster struck those areas, every U.S. hospital, pharmacy, and pediatric practice would feel the effects within weeks.
U.S. Pharmacopeia estimates that nearly seven hundred medicines approved for use in the United States depend on at least one chemical produced solely in China, with KSMs for drugs for statins, antibiotics, seizures, cancer, and HIV.19 Unless required to do so, companies are not likely to buy U.S.-made chemicals (or those from other markets) unless they are sold at China-like prices and scale. In 2023, a U.S. Department of Defense official testified that the national security risks of Chinese dominance of the global market for APIs “cannot be overstated . . . should China decide to limit or restrict the delivery of APIs to the United States, it would have a debilitating effect on U.S. domestic production and could result in severe shortages of pharmaceuticals for both domestic and military uses.”20
U.S. Pharmacopeia estimates that nearly seven hundred medicines approved for use in the United States depend on at least one chemical produced solely in China, with KSMs for drugs for statins, antibiotics, seizures, cancer, and HIV.
Over the last decade China has also surged up the innovation ladder to become a major force in the biotechnology sector via a coordinated state-wide campaign that has incorporated regulatory overhaul, subsidies, dedicated biotech hubs, and other forms of government support on a scale few other nations can match.21 The campaign began with regulatory reforms undertaken in response to a series of scandals involving corruption at China’s food and drug authority in 2007, contaminated Chinese-made heparin in 2008, and substandard rabies and diphtheria vaccines in 2018. These reforms improved the enforcement of bioequivalence for Chinese APIs and inputs and dramatically cut the approval time for clinical trial applications and new drug products, supporting China’s contract research industry that now conducts many of the early-stage development and first-in-human trials for multinational firms.22
It is a strategy that has capitalized on cumbersome FDA approval processes for early-stage clinical research, which shifted many phase I clinical trials abroad, mostly to China.23 China has also used joint venture requirements and conditioned marketing approval of drugs to compel foreign pharmaceutical firms to share their drug formulations, manufacturing processes, or research data with local firms and regulators. Heavy government support is helping BioMap, Baidu’s life sciences arm, and WuXi App Tec, China’s leading biomanufacturer, emerge as advanced biotech powers. In the first half of 2025 alone, U.S. pharmaceutical firms signed fourteen licensing agreements potentially worth $18.3 billion to restock their innovation pipelines with drug and vaccine candidates from Chinese biotech firms. Those agreements stand in contrast to just two such deals in the same period the year prior.24
A Brief History of the U.S. Response to Pharmaceutical Dependence on China
U.S. policymakers have been concerned for nearly two decades about U.S. dependence on China for active pharmaceutical ingredients and key starting materials for many critical medicines. Although the United States has changed its perception of the problem from a regulatory to a national security challenge, it continues to struggle to rectify the problem in any meaningful way. The figure below highlights certain milestones along that journey to date.
Pathways for Weaponization
Choke points in critical supply chains share three characteristics.25 First, a nation or coalition of allies has a dominant, concentrated market share in a critical input or production stage. Second, substitutes are not immediately available. Third, that country has a practical means of weaponizing its position in ways that create asymmetric leverage, inflicting substantial pain on the target while suffering minimal harm itself.
Pharmaceutical supply chains are particularly susceptible to choke points because both supply and demand for essential medicine inputs and production processes often do not readily respond to changes in price. That may be the case for the demand for an essential medicine if it is a medical necessity to patients and no good therapeutic substitute exists. The supply of an essential drug is similarly constrained, especially in the short term, because existing suppliers must obtain FDA approval for new manufacturing facilities or production lines needed to meet the shortfall. New entrants face similar regulatory obstacles and have to offer lower prices than existing producers to gain market share, while innovative production technologies often struggle to gain traction in a highly regulated sector that is understandably path-dependent and conservative.
Older U.S. generics are particularly susceptible to shortages because margins are low. A 2025 study found that once U.S. drug shortages occur, they tend to persist, lasting three years on average (up from two years in 2020).26 Shortages affect both purchasers—spurring hoarding and higher health-care costs—and patients, delaying surgeries and leading to less effective or less safe alternatives, medication errors, and avoidable deaths.27
In 2019, Chinese President Xi Jinping announced a “whole nation” effort to advance the biotechnology and pharmaceutical sector, deploying the full weight of the authoritarian state across central ministries, provincial governments, academia, and industry to pursue the nation’s strategic priorities in this sector.28 This broad authority empowers China with a wide range of options to exercise leverage over U.S. pharmaceutical supply chains.
China could weaponize supply cuts by imposing direct or indirect export restrictions. China has previously exercised its authority to require that exports of medical products—including COVID-19 testing kits, medical face masks and protective suits, ventilators, and infrared thermometers—be accompanied by proof of registration with the National Medical Products Administration. Alternatively, China could resort to more indirect, subtle regulatory measures that delay shipments or temporarily halt production at a critical facility, accomplishing the same practical effect of choking off needed supplies to U.S. patients and purchasers.
China can—and does—cut prices to disable competing supply chains. For generics, producers all along the global supply chain operate on thin margins due to robust price competition, reimbursement limits, or fixed contracts. Chinese producers recently flooded the market with below-cost exports of APIs and KSMs to cripple alternative suppliers in India that were supported under India’s Production-Linked Incentive scheme.29
A further risk is China weaponizing information. China need not impose a physical export restriction to create downstream stress; threatening restrictions or amplifying rumors via channels like TikTok can trigger anticipatory hoarding and panic-buying by consumers, hospitals, group purchasing organizations (GPOs), wholesalers, and manufacturers. In a buffered multitier supply chain, this behavior converts perceived scarcity into real scarcity by pulling forward demand, distorting order signals, and exhausting inventories faster than normal consumption would. The 2022 infant formula crisis illustrated how media attention and panic buying can intensify shortages beyond the underlying physical disruption. Similar behavior was observed during the COVID-19 pandemic, when hospitals and buyers over-ordered critical products in anticipation of future scarcity.
China could also pursue subtler forms of coercion by weaponizing quality, with Beijing’s manufacturers adulterating inputs or allowing manufacturing standards to quietly erode. Weaponization of quality—whether through deliberate adulteration or because of inadequate standards—represents a form of supply chain disruption particularly difficult to attribute and counter. The 2007–08 contamination of Chinese-manufactured heparin, which killed eighty-one Americans, illustrated that harm can propagate through supply chains even without formal export restrictions. Whether or not that particular episode was deliberate, the possibility that it could be repeated intentionally is a form of risk that cannot easily be deterred. As pharmaceutical inputs become more complex and AI-driven synthesis makes their origins harder to trace, quality becomes an increasingly viable instrument of quiet pressure. Quality problems create plausible deniability, enabling a pathway for weaponization that is not always straightforward and may remain deliberately obscured.
Thus, assessing the risk of China weaponizing the U.S. pharmaceutical supply chain usually boils down to situations where China has dominant, concentrated market share and where pathways exist through which China could practically leverage that dependence in a manner that adversely affects the United States. Four such scenarios exist.
Scenario 1: China restricts access to raw materials, KSMs, APIs, or FDFs that go from China directly to the United States.
China’s influence over U.S. pharmaceutical supply chains runs through two parallel paths: the inputs—raw materials, KSMs, and APIs—that provide the foundation for global manufacturing and finished dosage forms exported directly to U.S. patients. Currently, a relatively small percentage (an estimated 13 percent) of U.S. generic drugs and their inputs are imported directly from China, but this figure underplays the risk in certain vulnerable drugs that are highly dependent on Chinese manufacturers for finished dosage forms sold in the U.S. market.30
The oral solid products are dominated by antihypertensive medications—drugs taken daily by over fifty-five million Americans.31 Although they would not necessarily lead to an immediate crisis, shortages of these drugs would result in serious public health consequences. As multiple drugs in the same therapeutic class are included in this list from the same manufacturers, supply disruptions are unlikely to be isolated, leading to simultaneous shortages across related medications, thus limiting substitution options.
The U.S. injectable products imported directly from China include the immunosuppressant mycophenolate, which is critical for preventing organ transplant rejections; some broad-spectrum hospital antibiotics; and an acute heart failure agent. Again, the products are not easily substituted and represent the standard of care in some of the most common and serious conditions used in hospital settings. Mycophenolate is the standard of care in transplant surgery: a shortage would pressure clinicians toward alternative drugs with higher rejection rates and poorer graft outcomes.32 Cefepime is one of the most commonly reported antimicrobials to fall into shortage, with hospitals often shifting to less suitable drugs that carry downstream consequences for antimicrobial resistance and patient outcomes.33 Dobutamine is only one of two inotropes (drugs used in cases of cardiogenic shock and acute heart failure) accessible in the United States.34
Further, although the volume of direct U.S. imports from China in FDF is currently limited, it is poised to increase as China produces more innovative medicines on which the United States relies. Though the bulk of Chinese exports to the United States are in the form of KSMs and APIs, including indirect dependence from foreign manufacturers who source from China, the Chinese manufacturer is in many cases not the entity visible in the U.S. market. Rather, a U.S.-based company holds the drug label and distributes the product domestically but sources the API or the finished medication from a Chinese manufacturer under contract, sometimes without FDA authorization, such as with certain anti-obesity GLP-1 agonists. This structure can obscure the true origin of manufacturing when supply chain analyses rely on label holders’ market share rather than underlying producers. The FDA recognizes and inspects the underlying manufacturer as part of its oversight, but the arrangement means that the concentration of foreign manufacturing risk could be significantly understated in conventional market estimates.
Scenario 2: China restricts access to raw materials and KSMs to a third country on which the United States relies for imports of APIs or FDFs, and the United States suffers collateral damage.
Beyond direct imports, China’s upstream dominance creates an additional, less visible, channel of risk: restrictions on raw materials or KSMs shipping to India or Europe could trigger U.S. shortages collaterally, even in the absence of direct trade disruption between the United States and China. Nearly 41 percent of KSMs used in U.S.-approved APIs come solely from China and 16 percent from India.35 Accordingly, much of the supply of APIs and FDFs that the United States imports from India or Europe for antibiotics, analgesics, steroids, and other widely used essential medicines indirectly comes from China. If China were to leverage its market dominance in upstream supply against that third country, the United States would suffer shortages too. For instance, China recently imposed export controls against shipping dual-use products to Japan, after its prime minister gave public remarks on the Taiwan Strait.36 That list of dual-use products includes chemicals that are also used in pharmaceutical manufacturing, including solvents, reagents, fluorinated compounds, and certain fermentation-derived intermediates. If China imposed such a policy against India, it could result in serious consequences for the United States.
Because this approach would affect numerous countries and, potentially, a variety of medicines that incorporate that KSM, it would be a blunt policy instrument unlikely to align with China’s more targeted past behavior. Though some Chinese commentators have suggested leveraging the market share of Chinese API producers to serve Beijing’s policy goals, even its rare-earth critical mineral restrictions, in practice, included limited carve-outs for medical uses.37
Scenario 3: China restricts a third country from exporting API or FDF products to the United States made with raw materials or KSMs from China.
China could use global diplomatic pressure, regulatory measures, threats to cut off access to its market, and export controls to prevent companies and third countries from selling targeted KSMs, APIs, or FDFs to the United States. China has leveraged such export controls on critical minerals. The United States has deployed, with only limited success, related measures to prevent foreign government choke point suppliers and non-U.S. companies from exporting advanced chips and semiconductor manufacturing equipment to China. China could seek to do the same with pharmaceuticals, but it would require the cooperation of other producing governments and undoubtedly spur the same all-out push for alternative sources and self-sufficiency as those export measures in other sectors have.
Scenario 4: China restricts access to critical data or R&D infrastructure to U.S. drug developers and producers.
Increasingly, first-in-human clinical trial activity for promising medicines is shifting away from the United States and Europe to China, where those trials can be conducted faster and more cheaply. China’s share of global trial starts rose from 1 percent in 2009 to 30 percent in 2024.38 As China gains share in early stages trials and the resulting data grows, it creates a strategic dependence that could undermine U.S. leadership in translational science, and slowly choke off U.S. capacity to engage in preparedness and countermeasure development.
How China Weaponizing U.S. Pharmaceutical Dependence Could Play Out
The likely effect of a U.S. pharmaceutical supply disruption can be assessed across six dimensions: (1) how quickly the disruption reaches the market; (2) where the disruption occurs along the supply chain stages; (3) the average scale of shortages or cost increases; (4) the worst-case shortages at their peak once stockpiles are depleted and panic buying has amplified the problem; (5) the capacity to absorb the shock through excess inventory, alternative suppliers, or substitution; and (6) the time required to recover through approvals for new suppliers, restarted production, or expedited imports.
The severity of a disruption from China choking off supplies would depend on where in the supply chain it occurred. Restrictions at central, highly upstream points are more dangerous than later-stage supply shocks because they affect all downstream production paths. Therapeutic substitutability can reduce patient impact but can also shift demand to other antibiotics, leading to new shortages and creating secondary effects. Substituting sources at any tier of the supply chains (KSM, API, FDF) requires the FDA to assess and validate new batches, stability data, regulatory filing updates, and inspection readiness; buyers cannot simply switch suppliers without FDA approval. A CFR-New England Journal of Medicine analysis shows temporary importation can mitigate certain generic shortages, but the FDA generally deploys it only after other options are exhausted, even after months of delay. In one benzathine penicillin G case, nearly nine months passed before FDA authorization.39
Excess inventory and time buffers could delay the impact of China-instigated supply chain disruptions but in turn create a false sense of stability and surprise effects. Contract rigidity affects whether a shock manifests as higher prices, margin compression, or allocation. Typical human behavior in a crisis—over-ordering, hoarding, inflated forecasts, rationing—further accelerates propagation.
Most scenarios short of complete supply shutoff usually manifest first as price increases rather than immediate shortages: upstream inputs become more expensive, scarce suppliers raise quotes, buyers bid up available volumes, and downstream firms draw down inventory while repricing. In generics, price sensitivity runs through the entire chain: API manufacturers, FDF manufacturers, wholesalers, pharmacies, hospitals, and payers often operate under competitive pricing, tendering, reimbursement limits, or fixed contracts. As a result, upstream cost inflation may not fully pass through, instead appearing as margin compression, delayed replenishment of supplies, or even the exit of manufacturers. For innovative medicines and biologics, product pricing is largely decoupled from cost of goods sold (driven by patent protection, market access, contracting, and payer dynamics), so upstream input shocks are more likely absorbed as margin impact or supply-risk management before becoming visible price changes.
China controls about 94 percent of 6-APA (the main KSM for amoxicillin); API manufacturing occurs in Austria, China, and India; FDF manufacturing occurs in India, the European Union, and sometimes in the United States. A simultaneous Chinese restriction on both 6-APA and amoxicillin API hits two tiers at once: non-China API manufacturers lose KSM access while FDF manufacturers lose both direct Chinese API and, over time, non-Chinese API. Under a 25 percent restriction, effective global API availability falls to roughly 80 percent (around a 20 percent deficit); under a 50 percent restriction, availability falls to roughly 60 percent (around a 40 percent deficit); and under a full ban, availability falls to about 20 percent, essentially Austria plus a small residual (around a 79–80 percent deficit). Because API manufacturers normally hold six months of KSM inventory, FDF manufacturers hold three to six months of API inventory, wholesalers hold thirty days, and pharmacies hold roughly fifteen days, the first signals would be API price increases, allocation, delayed replenishment, and tender failures—not pharmacy-level shortages. Once buffers deplete, a threshold effect drives sudden acceleration: peak shortages would arrive roughly within 6 to 9 months after a 25 percent restriction, 5.5 to 8.5 months after a 50 percent restriction, and 4.5 to 7.5 months after a full ban, with hoarding and over-ordering capable of pulling all timelines forward.40
An Archetype Approach to Understanding U.S. Pharmaceutical Dependence
This report aims to create a replicable analytical policy framework—the archetype model—for policymakers, agencies, and researchers to apply to other product classes that share the same root causes of dependence on China. A one-size-fits-all approach won’t work for redressing the “rare-earths’ problem” in the U.S. pharmaceutical supply chain. The root causes of dependence often differ by product class and can arise at different stages of the value chain. Policy solutions must target those particular differences to be effective. Upstream market concentration in KSMs would only be masked by downstream supply diversification. The perilously low margins for older generic pharmaceuticals create sterile manufacturing facility fragility that cannot be redressed by re-shoring or sixty-day stockpiles alone. Country-of-origin labeling (COOL) or Buy America approaches need to account for transshipment, domestic branding, and API repackaging. R&D infrastructure capture reflects a systemic competitive erosion via Chinese state-backed integration and coordination that can only be addressed in kind.
Archetype One: Reliance on China for Raw Materials and Upstream Supplies for Generic Drugs
The defining characteristic of this archetype is a choke point at the raw material or key starting material stage and at the intermediate level. This archetype is defined by the “N-1 problem”: apparent diversification at one stage often masks near-total concentration at upstream inputs where true dependence has quietly shifted up one critical step, the “minus 1.” When all downstream suppliers depend on that same concentrated input, downstream diversification (at the level of APIs or FDFs) provides no protection. It is an end-to-end supply chain challenge, and policy responses need to treat it as such, by mapping upstream tiers and arranging geographic diversification of starting material sourcing with allies.
Most medicines in the first archetype share a structural vulnerability. As low-cost but essential generics, these medicines no longer generate sufficient economic returns to justify domestic vertical integration. U.S. producers face structural cost disadvantages from environmental compliance, while GPOs concentrate buying power and drive a race to the bottom on price—functioning as embedded subsidies for foreign production. Partial investments at the API or FDF level, without corresponding KSM capacity, leave the core vulnerability intact. Total independence from foreign sources would require an end-to-end domestic supply chain: a solution demanding high capital, lead times of five to ten years, and sustained policy commitment that market forces alone cannot produce. This dynamic—low-margin generics, GPO price pressure, and shortage costs externalized onto patients rather than purchasers—recurs across our four case studies in Archetype One.
These case studies all reflect the broader trend of supply risk moving further upstream, where dependence on essential raw materials or intermediates has become a critical vulnerability. The nature of that upstream risk varies, however: certain inputs are synthetically produced, such as fermentation-derived materials, while others are naturally harvested, such as porcine mucosa (from which heparin is derived). The cases also differ in regulatory and manufacturing complexity, including whether they involve combination APIs under Drug Enforcement Administration (DEA) oversight and whether production depends on sterile injectable capabilities versus more flexible oral solid manufacturing.
Case Study: Amoxicillin
Amoxicillin is the most prescribed antibiotic in the United States, with nearly sixty million prescriptions per year, treating infections across hospitals, pharmacies, and clinics.41 It is especially critical in pediatric care and for seniors over sixty-five, who use it at rates nearly 50 percent higher than younger Americans and depend on it for routine procedures such as hip replacements and cancer surgeries.42
Why This Product Is a National Security Concern
As recently as 2008, nearly all U.S.-administered amoxicillin was domestically manufactured.43 Subsidized competition from Chinese and Indian generic manufacturers drove U.S. producers from 100 percent market share to 0 percent within twelve years.44 Amoxicillin now sits on both the FDA and World Health Organization (WHO) emergency medicines lists, signifying that its uninterrupted supply is considered essential to basic health-care capacity.45
As recently as 2008, nearly all U.S.-administered amoxicillin was domestically manufactured. Subsidized competition from Chinese and Indian generic manufacturers drove U.S. producers from 100 percent market share to 0 within twelve years.
Without a reliable supply, cases as simple as minor wound infections can become life-threatening. The 2022 amoxicillin shortage—triggered by an ordinary demand surge in peacetime conditions—resulted in a 30.8 percent decrease in amoxicillin prescriptions in U.S. pediatric hospitals, accompanied by an increase in broader-spectrum antibiotics that accelerate antimicrobial resistance.46 More broadly, the shortages led to documented cases of rationing, delayed surgeries, and substitution with less effective products. Those shortages offer a clear stress test: U.S. supply chains failed under everyday conditions, thus demonstrating that even minor disruptions can be devastating.
Structural Vulnerability
Although the API and FDF layers of the U.S. amoxicillin supply chain appear geographically distributed, they are all anchored to a single upstream choke point: penicillin G and its derivative 6-APA. Nearly all U.S. supplies of penicillin G originate in China, with near-zero Indian sourcing, making intermediate 6-APA stages dependent on this same base.47 Five Chinese companies, including the state-owned North China Pharmaceutical Corporation, control over four-fifths of global 6-APA production. In parallel, our own analysis shows China supplying 94 percent of global amoxicillin starting materials; India, dominant in amoxicillin FDF manufacturing at 59 percent, sources its intermediates from China as well. The result is American pharmacy shelves stocked with the products of Chinese chemistry with Indian finishing—a supply so compressed that any upstream disruption propagates to American consumers within weeks. The starting materials of API manufacturers in Europe (Austria and Spain) and Jordan are also traced back to China, meaning even non-Chinese producers of amoxicillin are essentially reliant on Chinese inputs. Diversification at the API and FDF stages therefore fails to provide meaningful protection against Chinese supply disruption—but it does create a dynamic in which China would need to leverage against the United States with the cooperation of another country, such as India, or as part of a larger effort against nations that rely on amoxicillin made with key starting materials from China.
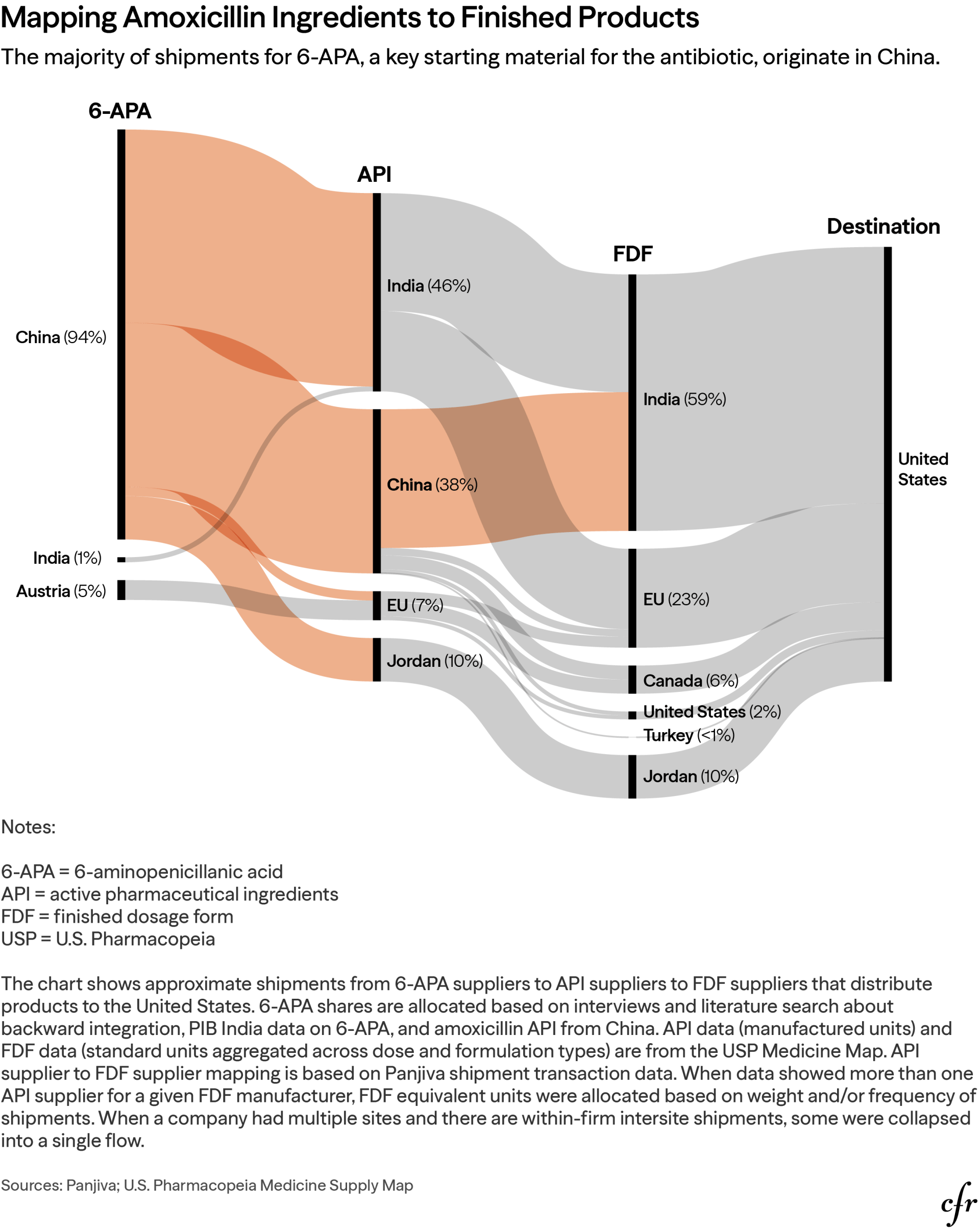
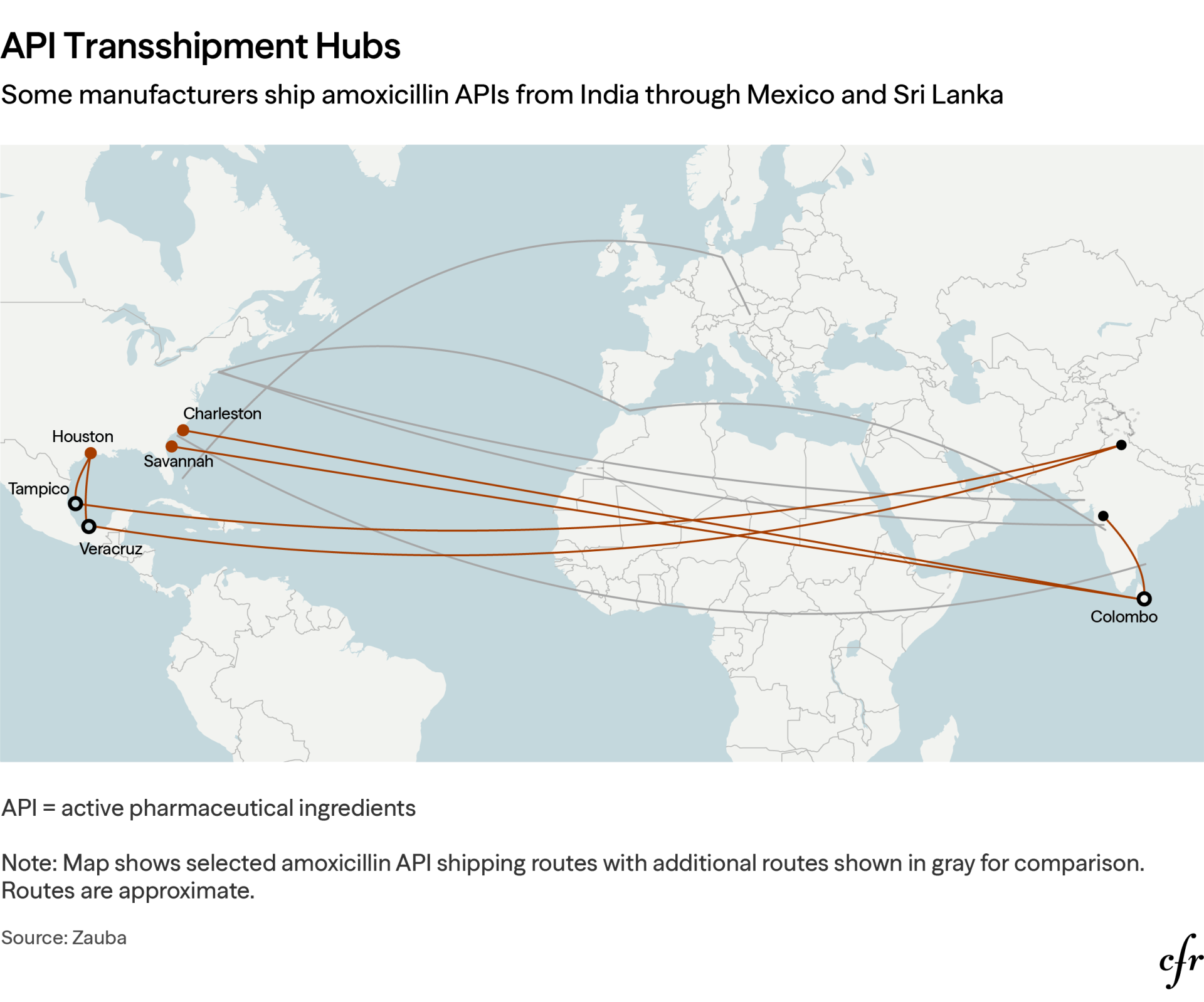
Trade and customs data reveal that certain Indian manufacturers route APIs through transshipment hubs in Mexico and Sri Lanka before entering U.S. ports, which obscures the true origin of the supplies and undermines country-of-origin enforcement. Those diversions disable current customs and regulatory oversight, blinding officials to the true supply chain concentration risks, while funneling multiple nominally independent supply lines through a handful of southeastern U.S. ports—creating a physical choke point mirroring the upstream chemical one.
Case Study: Heparin
Heparin represents the clearest case where the choke point has shifted fully upstream to the starting material stage, presenting the strongest raw material concentration risk in this report. The drug is based on a biological input with sourcing overwhelmingly from China, no meaningful domestic alternative, and a recorded history of intentional contamination by Chinese producers.48 For decades, heparin, a blood thinner, was made with bovine ingredients until the mad cow disease scare in the 1990s prompted manufacturers and regulators to adopt porcine alternatives instead.49 With that change, China, the world’s largest pork producer, also became the dominant supplier of crude heparin, now made only from pigs. Downstream diversification is irrelevant given global supply chains converge at the same biological input from China. Although the United States is the third-largest pork producer, it lacks the collection, processing, and transportation infrastructure needed to turn porcine mucosa, a byproduct, into a reliable domestic heparin input.50 Because API and FDF manufacturers already have access to cheaper established sources in China, they have little market incentive to make the capital investments required to build this infrastructure.
Why This Product Is a National Security Concern
Heparin, like amoxicillin, is one of the backbones of modern medicine. Heparin is administered to more than 12 million hospitalized Americans per year, with approximately 265 million total doses provided annually.51 Appearing on every critical medicines list of relevant agencies—the WHO, the Department of Health and Human Services (HHS), the FDA, and the Department of Defense (DOD)—its clinical span is unusually broad, spanning common surgeries, kidney dialysis, asthma treatment, battlefield injuries, and biological countermeasures.52 Supply disruption would not just affect a single disease population but would degrade the operational capacity of hospitals nationwide. As of May 2025, the American Pharmacists Association lists heparin on its drug shortage list, with experts advising clinicians to begin rationing—a position reflecting baseline conditions, not a crisis scenario.53
Several times, the dependence on China for heparin has put the millions of U.S. patients who use the drug annually at risk. The 2019 African swine fever outbreak devastated China’s pig population and triggered a global shortage of heparin. A 2008 contamination crisis, in which adulterated Chinese-manufactured heparin API triggered a massive FDA recall, resulted in numerous deaths worldwide—including eighty-one in the United States.54 A supply disruption from 2017’s Hurricane Maria produced a 152 percent increase in medication error rates and a 114 percent increase in error rates for its substitute drug, enoxaparin.55
Structural Vulnerability
Heparin is derived from porcine intestinal mucosa, with 80 percent of the global supply and 74 percent of U.S. supply originating in China. Domestic collection accounts for less than 10 percent of global supply.56 The United States is the third-largest pork producer, but it does not produce porcine mucosa at meaningful scale for pharmaceutical use.57 Porcine mucosa is a slaughter byproduct that requires dedicated collection, segregation, preservation, and processing infrastructure to convert it into a pharmaceutical-grade heparin input. A regulatory decision made roughly twenty years ago prohibiting bovine-origin heparin over mad cow disease concerns foreclosed the primary pathway to geographic diversification.58 The U.S. Strategic National Stockpile has limited heparin holdings; supply disruptions translate into immediate clinical impact with no buffer. Indian producers such as Dr. Reddy’s Laboratories and multinational companies with significant Indian manufacturing, such as Mylan, source their API entirely from China. Hepalink—including its 2013 acquisition of Scientific Protein Laboratories in Wisconsin—has vertically integrated from mucosa collection through finished product, cementing Chinese control across the full value chain.59
Regulatory inertia and a lack of explicit incentives from FDA to switch to bovine heparin suggest that no private actor will invest in rebuilding domestic mucosa collection and bovine processing infrastructure. But Brazil’s approach—establishing separate standards for porcine and bovine heparin in 2019—demonstrates that this is a manageable regulatory problem rather than an intractable market barrier.60
Case Study: Norepinephrine
Norepinephrine is the foundation of resuscitation medicine, and shortages of it create a vulnerability that is simultaneously a civilian health-care crisis and a military readiness concern. Norepinephrine, which can treat life-threatening low blood pressure and sudden shock, is used in the vast majority of U.S. hospital trauma cases, is dependent on foreign API, and is constrained by aging domestic sterile injectable manufacturing. COVID-era shortages have already demonstrated system failure under stress without any adversarial trigger.61
Why This Product Is a National Security Concern
Norepinephrine is essential across military field care, civilian emergency rooms, intensive care units, and operating rooms, and is core to cardiac arrest algorithms. It is one of the most operationally critical hospital drugs, and supply disruption would simultaneously impact the broadest and most acute ends of the patient care spectrum. It appears on essential medicine lists from the DOD, the Administration for Strategic Preparedness and Response (ASPR), and the FDA.62
Shortages translate directly into preventable deaths: during the 2011 U.S. norepinephrine shortage, a study confirmed a 3.7 percent increase of in-hospital mortality among septic shock patients.63 COVID-era shortages demonstrated how easily the supply chain fails under demand stress even without the prompting of an adversary, and instead can be strained by fragile just-in-time inventories and opaque global sourcing.
Structural Vulnerability
Norepinephrine’s core vulnerability stems from two compounding factors that cannot be independently addressed: dependence on foreign API sources that, in turn, rely on KSMs from China, and domestic sterile injectable manufacturing capacity constraints. Resolving one without the other still leaves the supply chain exposed.
Sterile injectable production requires capital-intensive high-precision aseptic manufacturing, yet capable U.S. facilities for manufacturing this low-cost generic are aging and operating on margins too thin to support modernization or expansion. Robust competition keeps the prices for norepinephrine low, creating no commercial incentive to invest in the quality or resilient supply beyond what is needed for FDA minimum standards. The just-in-time production and inventory model for norepinephrine is a structural consequence of margin pressure, not an operational choice that individual actors can unwind absent policy intervention. Minimal redundancy exists in the domestic manufacturing base, and even a single plant failure can collapse national availability.64 Those manufacturing constraints are compounded upstream: heavy reliance on foreign APIs from regions vulnerable to coercion from China for their upstream KSM supplies means that even a functioning U.S. fill-finish operation remains exposed to input disruption.
Case Study: Acetaminophen
Acetaminophen, the most widely used analgesic in the United States, shares the upstream concentration risk of the cases above but has one important difference: separate over-the-counter (OTC) and prescription drug markets that together obscure the extent of U.S. dependence on China.
Why This Product Is a National Security Concern
Used by fifty-two million Americans every week and on the emergency medicines list for the HHS, FDA, and WHO, acetaminophen’s scale of dependence has no true equal among the other products in this study.65 Broad OTC access serves a preventative function—keeping patients with mild-to-moderate pain and fever out of emergency rooms—meaning supply disruption would generate downstream pressure on health-care infrastructure, not merely inconvenience consumers. It is consistently stocked in military field hospitals and deployed medical units, critical for troop readiness, and embedded in multimodal pain management protocols combining it with opioids across civilian and military surgical and trauma care.
Structural Vulnerability
Although domestic finished-product manufacturing creates the appearance of resilience, 55 percent of global acetaminophen API comes from China and relies on a small number of primarily Indian FDF manufacturers.66 Meanwhile, the supplies for the higher-cost prescription version of acetaminophen are made domestically, again creating the perception of U.S. resilience. As acetaminophen is sold predominantly as an OTC product under domestic brand names, the fragility of its supply chain is masked by normal market mechanisms. Retailers see competitive prices, consumers see familiar labels, and the concentration risk remains unpriced and uncorrected. Like amoxicillin and norepinephrine, the OTC market structure provides no commercial incentive to correct this vulnerability: high-volume private-label demand from major retail chains, funneled through GPOs, suppresses margins throughout the supply chain.
In contrast, according to USP Medicine Supply Map data, the API market for higher-margin prescription opioid-combination formulations (e.g., acetaminophen/codeine) is almost entirely U.S.-based, as the figure below shows. This market segment, however, represents only a small fraction of total acetaminophen demand. Whether these domestic API suppliers could absorb broader market demand under a stress scenario remains unclear, creating a critical surge capacity gap with no clear corrective mechanism. Switching from one API supplier to another is not straightforward because the particle-size distribution for API for pediatric formulations is different than other versions.
An Industrial Policy for Archetype One: Near-Term Management of Acute Risks Combined With Longer-Term Market and Supply Creation
A credible Archetype One industrial policy—addressing choke points at the raw material or KSM level on which all downstream suppliers depend—needs to combine near-term risk management with integrated longer-term market and supply creation. The goal is targeted resilience: minimizing China’s opportunity to extract strategic gains from weaponizing U.S. supply chains rather than reshoring all production or replicating the China model.
Without also addressing upstream KSM, excipients, and intermediates, onshoring API alone will not significantly reduce the China risk to Archetype One medicines like amoxicillin or heparin. Equally, securing upstream inputs will not prevent shortages absent excess aseptic fill-and-finish capacity to transform those inputs into sterile injectables such as norepinephrine. Eliminating the full end-to-end risk of a China shock requires coordination across KSMs, intermediates, API, and finished product—sustained by a multiyear industrial policy that outlasts election cycles.
The goal of an Archetype One policy is targeted resilience: minimizing China’s opportunity to extract strategic gains from weaponizing U.S. supply chains rather than reshoring all production or replicating the China model.
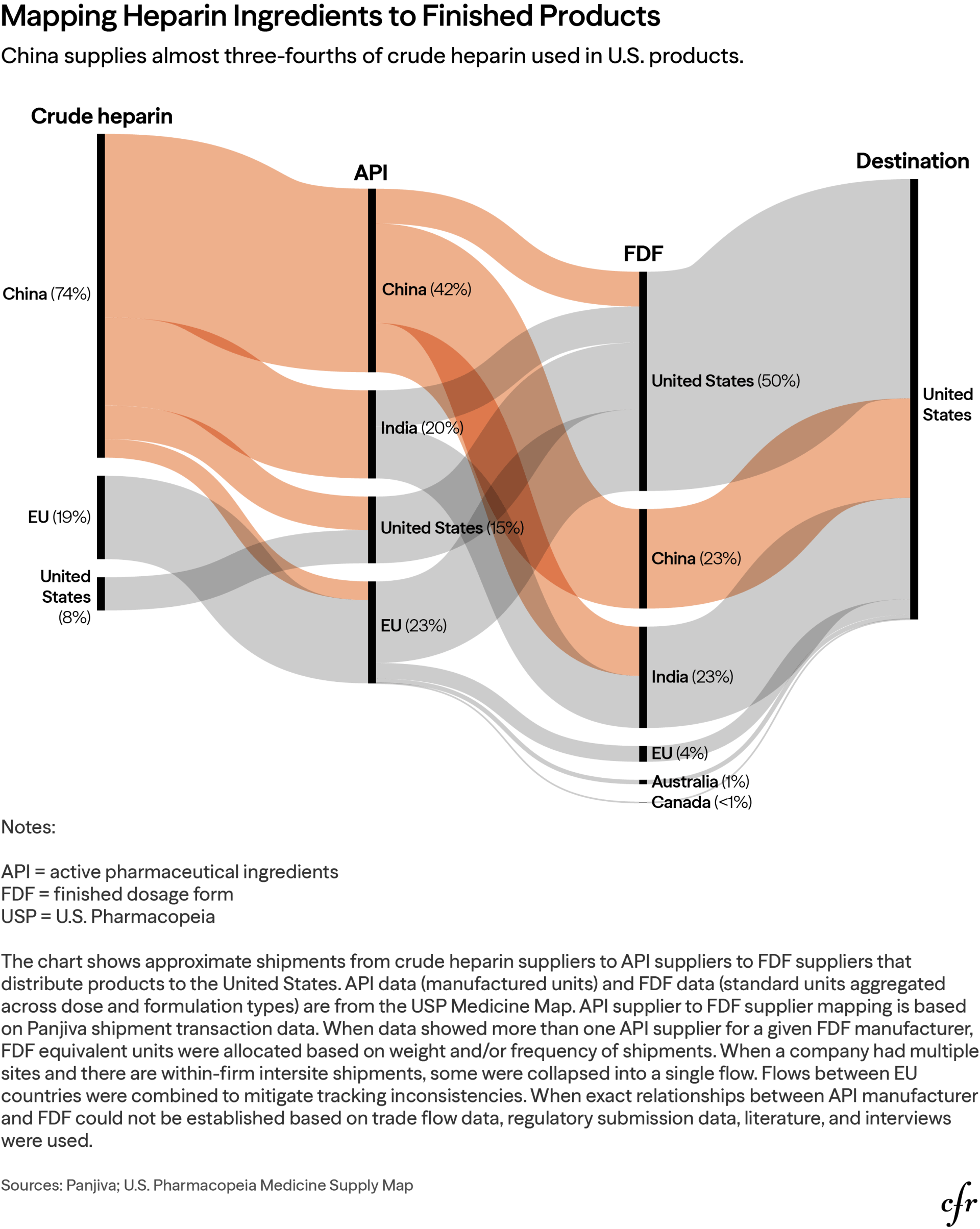
Priority should go to essential medicines imported directly from China and where China holds concentrated market dominance, as those products and inputs are most susceptible to weaponization. Our analysis suggests, for example, that the United States depends on imports from China for mycophenolate mofetil, dobutamine, and cefepime, which appear on multiple U.S. essential medicines lists and the essential medicines list for the WHO.67 Heparin also warrants priority: the U.S. imports nearly a quarter of its FDF supply from China and relies on China for 42 percent of its API and three-quarters of the crude heparin used in U.S. medicines.
Amoxicillin, acetaminophen, and norepinephrine are examples of medicines appearing on both U.S. and WHO essential medicine lists where China dominates upstream inputs, but whose supply is exported through a third country rather than directly to the United States.68 Leveraging this exposure requires China to entice or compel that third country—typically India. This might occur when a supply shock in China restricts KSM and other inputs for essential medicines and the Indian government prioritizes its domestic population, as India did during COVID-19.69
Near-Term Management of Acute Risks
Over the next two years, the federal government should use the Strategic Active Pharmaceutical Ingredients Reserve (SAPIR) or a similar strategic reserve mechanism to stockpile a minimum six-month supply of critical KSMs and APIs most at risk of weaponization from China. At the same time, the United States should enter into arrangements with FDA-approved allied (non-China) API suppliers for each of those medicines and have a clearly defined expedited pathway for temporary importation to improve buffer capacity. Finally, the United States should establish risk communication protocols for essential medicine shortages to reduce panic and hoarding by states, hospitals and other institutional purchasers, and patients.
Create a functional ecosystem for a strategic reserve of KSMs and APIs for priority critical medicines. Stockpiling has been explored by the United States at two stages of the pharmaceutical supply chain: FDF and API. FDF stockpiling, as practiced in the Strategic National Stockpile, enables immediate deployment during a crisis without downstream manufacturing, benefiting from preexisting regulatory approvals. Drawbacks are significant, however, including shorter shelf lives (one to five years), higher storage and cold-chain costs, limited formulation flexibility, and a design oriented toward health emergencies rather than weaponization scenarios.
SAPIR was launched in 2020 in response to the COVID-19 pandemic. It has promise: APIs and KSMs usually have longer shelf lives and lower storage costs than FDFs and can be converted into multiple FDFs when necessary. ASPR, which oversees the SAPIR, was intended to identify at-risk APIs and stockpile critical ingredients in U.S.-based GMP-compliant facilities. To convert those APIs into FDFs, it relied on a distributed network of sterile fill-and-finish facilities where FDFs were packaged for distribution. Instead, SAPIR languished largely unfilled and underfunded during the Biden administration.70 The Trump administration is seeking to revitalize stockpiling active ingredients for some top priority medicines, but without a supporting ecosystem, this effort will fall well short of more expansive resilience goals.71
The SAPIR depends critically on an FDA-approved finished-dosage manufacturing network positioned to rapidly convert APIs into labeled medicines—with all regulatory approvals already in place. Certain manufacturers will also require liability protection before using stockpiled APIs and KSMs outside their control.
True resilience requires a layered approach: API reserves combined with formulation capacity and targeted FDF buffers. Stockpiling protects against disruption but cannot resolve the underlying structural concentration that makes weaponization possible—only supply diversification can.
Congress should direct and appropriate funds for ASPR to purchase at least an eighteen-month supply of essential KSMs and APIs for the existing SAPIR facility under a dynamic life-cycle rotation program.72 ASPR, working with the FDA and HHS, should then coordinate and fund a distributed network of aseptic filling sites—equipped with robotics, isolator technology, and single-use systems—capable of rapidly converting stockpiled APIs into finished drug products during shortages or emergencies.
Create allied coordination mechanisms for KSM production to distribute geographic risk, including regulatory cooperation. API manufacturing is largely offshore, and not just in China. The United States should leverage alliances to reduce shared pharmaceutical dependence on China—preventing shortages and limiting China’s ability to weaponize supply chains through third countries. Allies should equally be able to rely on U.S. strategic posture as part of their own pharmaceutical supply chains.
The United States should facilitate and rely on cost-competitive allied API production rather than seeking to replicate China’s full capacity domestically. Fortunately, some allied governments are already taking steps to reduce their dependence on China. The European Union, under the Critical Medicines Act, has expedited approvals for manufacturing certain medicines dependent on Chinese supply chains.73 Japan is restarting and upgrading its production of critical antibiotics produced via fermentation.74 India has recently launched incentive schemes to build domestic capacity for KSM, intermediates, and APIs.75
In February 2026, the Office of the U.S. Trade Representative (USTR) sought comments on a plurilateral critical minerals trade bloc offering preferential terms to partners and discriminating against Chinese-processed material. A similar arrangement, paired with mutual recognition agreements, could enable allied sharing and purchasing of critical KSM and API stockpiles.76
Finally, the FDA should more readily authorize temporary API importation from well-regulated allied markets. Although that is already permitted, the FDA only does so as a last resort after months or even years of delay. The agency should proactively identify and assess foreign sources for essential medicines where China holds a concentrated market dominance.
Risk communication plans are part of U.S. supply chain resilience. The ASPR, the Centers for Disease Control and Prevention (CDC), and state and local public health agencies need pre-positioned plans for communicating essential drug shortages to the public, hospitals, and clinicians to prevent the panic buying and misinformation that may itself be China’s coercive goal.77 The National Academies’ report on the 2022 infant formula shortage affirmed that communication and expectation setting are core components of supply chain resilience.
Longer-Term Demand-Side Interventions
The U.S. federal government has tools, such as the Department of Health and Human Services’ (HHS) Defense Production Act (DPA) Title III Program, to subsidize the resiliency, diversity, and security of supply chains and to offer grants, tax incentives, loans, and loan guarantees for domestic manufacturing of many key pharmaceutical inputs. Some states have sought to go further and proposed directly manufacturing generic drugs.78 But new or expanded capacity resulting from public investments may not continue long term as market demand for the product changes. For example, a company that received Title III funds to boost production of medical swabs for COVID-19 testing closed manufacturing sites in 2023 after a drop in demand.79
Over the next three to five years, policymakers should pursue a coordinated mix of longer-term demand and supply interventions—not just isolated fixes—to make domestic or allied production of KSM, API, and other upstream inputs economically viable and sustainable. The goal of those medium-term efforts should be to maintain at least 25 percent non-China KSM sourcing for essential medicines and inputs, such as amoxicillin (6-APA), heparin (crude mucosa), and norepinephrine API, and verify that sourcing through mandatory country-of-origin disclosure to FDA.
Require targeted API and KSM sourcing disclosure and labeling. The existing U.S. regulatory framework was not designed for upstream KSM or API transparency. The FDA requires only the name and place of business of the manufacturer, packer, or distributor on drug labels—not manufacturing locations—and officials acknowledge the agency cannot precisely determine China’s actual API export volumes.80
U.S. agencies have even less information on KSM manufacturers. The Drug Supply Chain Security Act of 2013 focused on finished-dose traceability, not upstream sourcing. Trade data lacks the granularity of customs bills of materials, which have some of the proprietary information companies closely guard. Data sharing even among fellow HHS agencies on drug sourcing and utilization is constrained. Defense Production Act surveys provide only point-in-time snapshots, not the dynamic visibility that supply chain management requires.81
Visibility into manufacturing locations is essential to supply chain risk management.82 Without it, hospitals, clinics, and retail pharmacies cannot know if a drug is made in a flood-prone location, a country with a history of export restrictions, or a facility with previous FDA quality violations, limiting their ability to anticipate other supply vulnerabilities, not just the threat of Chinese coercion.
Visibility into manufacturing locations is essential to supply chain risk management. Without it, hospitals, clinics, and retail pharmacies cannot know if a drug is made in a flood-prone location, a country with a history of export restrictions, or a facility with previous FDA quality violations, limiting their ability to anticipate other supply vulnerabilities, not just the threat of Chinese coercion.
Numerous stakeholders have recommended mandatory COOL for APIs on retail and wholesale labels.83 A new U.S. Customs and Border Protection (CBP) directive now requires pharmacy-repackaged drugs sold to retail customers to bear COOL markings and importers to certify they will notify pharmacies of their requirements.84 Implementation, however, remains contested: pharmacy associations argue that dynamic API and KSM sourcing makes labeling impractical. Legislation pending in Congress would extend COOL requirements to all U.S.-sold pharmaceuticals—FDFs and APIs—across the full supply and distribution chain.85
Those barriers are not insurmountable. New Zealand maintains a functional public national pharmaceutical supply chain data system. A phased, tiered approach offers a workable path forward, beginning with confidential, product-specific mandatory disclosure to regulators for essential medicines and then building toward aggregate public reporting and labeling consistent with BIOSECURE Act compliance requirements.
Bolster KSM- and API-level domestic investment, with well-defined Buy America terms that account for transshipment. Buy America provisions through Veterans Affairs (VA), the DOD, and Medicaid could create stable baseline demand for domestic and trusted allied APIs and KSMs. Medicare Part B should help offset higher U.S. manufacturing costs through more generous hospital reimbursement rates for domestically sourced products. Medicare Part B offering a premium reimbursement for domestically sourced medicines and inputs could have broader impact, with commercial payers and insurers using Medicare as a benchmark in determining reimbursement standards. The Centers for Medicare and Medicaid Services (CMS) used this model for N95 respirators, with additional payments for domestically manufactured products approved by the National Institute for Occupational Safety and Health, although burdensome requirements undermined uptake. A version of that basic model should be adapted for KSMs and APIs.
Defining and verifying whether the output is “U.S.-manufactured” has been the central difficulty of using volume guarantees, Buy America provisions, and price premiums from the DOD, VA, and CMS to create predictable demand for domestically sourced APIs. A 2023 DOD report found that 54 percent of DOD-sourced national drug code (NDC) medicines were noncompliant with the Trade Agreement Act, actually derived from China, India, or unknown origins via transshipment and repackaging.86 Policymakers must create clear standards for required shares of domestic API production and FDF manufacturing, with reliable CMS, hospital, and GPO tracking mechanisms to determine eligibility for additional payment. Extending preferences to trusted allied sources in well-regulated markets is also advisable, in part because shifting to an exclusively Buy America provision risks near-term shortages.
CMS could also preference Medicare Part B reimbursement of domestically sourced porcine or bovine API to offset import cost disadvantages. Expanding U.S. porcine mucosa collection would require USDA and FDA coordination on slaughterhouse practices, animal byproduct handling, traceability, and pharmaceutical-quality requirements, with subsidized capital for collection and preprocessing infrastructure. Funding from the Defense Advanced Research Projects Agency (DARPA), Biomedical Advanced Research and Development Authority (BARDA), Advanced Research Projects Agency for Health (ARPA-H), National Institutes of Health (NIH), National Science Foundation, and FDA could simultaneously accelerate new sources of synthetic and bovine heparin. In strategic cases, the federal government could consider equity purchases or direct investment in domestic heparin manufacturers.
Longer-Term Supply-Side Interventions
Target pharmaceutical tariffs on medicines containing APIs and KSMs from China. A tariff targeting products containing Chinese-sourced KSMs and APIs would be administratively difficult but would capture the actual points of dominance and national security risk, incentivizing production shifts to lower-cost allied sources.87 In contrast, blanket tariffs on imports of generics would only worsen U.S. pharmaceutical insecurity, disrupting supplies of essential chemotherapy agents, penicillin, and sterile injectable medications that are low-margin, complex to make, and hard to reprice under Medicaid, Medicare, and long-term hospital contracts.
Support investment in advanced manufacturing technologies and U.S. production. KSM and API production will not sustainably attract private investment to the United States or trusted allies without cost competitiveness at scale and greater demand certainty. Advanced manufacturing technologies (AMTs)—including continuous flow manufacturing (which uses automated processes to cut down on production time) and green chemistry (which lowers environment costs by using biocatalysts)—already exist and can improve reliability, quality, and cost efficiency. Yet manufacturers resist adopting AMTs for low-margin generics because of their up-front cost and regulatory uncertainty. The U.S. government should deploy coordinated public R&D investment, tax credits, and push-pull regulatory incentives to spur that adoption, prioritizing projects with measurable improvements in yield, quality, and energy efficiency. NIH, BARDA, and ARPA-H funding can provide the needed support for scaling up capital-intensive fermentation. Novel precursors such as ARPA-H’s WHEAT initiative have demonstrated the feasibility of this approach.88 Public-private partnerships could help introduce AI-driven process optimization and predictive analytics to improve API and KSM production efficiency and identify potential quality issues proactively.
In 2022, Congress authorized an advanced manufacturing technology program, enabling the FDA to designate technologies outside product applications and provide expedited review incentives for adopters. Uptake, however, has been limited. The FDA should clarify how regulatory frameworks designed for conventional manufacturing should be applied to AMTs and update AMT designation guidance to expand incentives, reducing regulatory uncertainty as a push to adoption.
Extended market exclusivity is a pull incentive that improves long-term returns for higher-cost domestic manufacturing; that is, advance purchase commitments reduce demand uncertainty for fermentation-based facilities. The FDA’s National Priority Voucher pilot, recently applied to Augmentin extended-release capsules for a U.S. manufacturer, offers a potential model, though its real-world impact remains to be assessed.89
Other risk-sharing mechanisms include multiyear tax credits for API and biologics facility investment, subsidized AMT equipment financing, and accelerated depreciation for pharmaceutical infrastructure. Sandoz’s fully integrated KSM-to-FDF plant in Kundl, Austria, proves coordinated action can resolve dependence on Chinese sources. A modest 50 million euro grant from the EU enabled enzymatic synthesis upgrades at this facility, making it cost-competitive with Chinese 6-APA and API sources.90
Expand production to lower-cost, more reliable allies and leverage API and KSM-incentive programs in Japan and Europe. The U.S. International Development Finance Corporation (DFC) and the Export-Import Bank should extend low-interest financing to projects that build API and KSM capacity in lower-cost allied nations. Geographic diversification of the U.S. supply chain should explore friendshoring partnerships with Argentina, Brazil, and Mexico. Within five years, the ASPR, DFC, and FDA should collaborate on the goal of completing qualification of at least one allied-nation KSM/API supplier for each U.S. priority essential medicine. Within a decade, the U.S. goal should be ensuring that there is domestic or allied production capable of meeting at least 50 percent of the normal U.S. demand for priority products’ KSMs or APIs.
Archetype Two: Erosion of U.S. Biopharmaceutical Manufacturing and Clinical Trial Capacity to China
The defining characteristic of Archetype Two is systemic competitive displacement of the United States across the full value chain for key innovative biotechnology products—R&D, clinical development, contract research and data management organization services, and commercial manufacturing—enabled by state-backed industrial policy at a scale and speed that U.S. market forces cannot match. These products require a “whole of value chain” industrial strategy, including robust federal investment in foundational R&D.
Case Study: Monoclonal Antibodies
Monoclonal antibodies (mAbs) are an innovative product class deployed in cancer immunotherapy, autoimmune treatment, and antiviral therapies, with a global market exceeding $380 billion.91 Monoclonal antibodies represent the case study of this archetype: emerging dependence on China where the vulnerability is most advanced and the trajectory most alarming. The value chain does not suffer from a specific upstream choke point but rather systemic competitive erosion across every stage—discovery, development, manufacturing, and market access—enabled by state-backed Chinese industrial policy operating at a scale and speed that U.S. market forces and fragmented government policy have not matched.
Why This Product Is a National Security Concern
Beyond their important everyday uses, mAbs are employed in response to pandemics and biological threats, including Ebola and anthrax. Supply disruption of inputs and finished mAbs products would cascade across most critical hospital treatments simultaneously. The COVID-19 pandemic previewed this fragility when capacity constraints forced direct government intervention to secure raw bioreactor materials and single-use components.92 New manufacturing facilities for mAbs require up to eight years to build, making domestic recovery impossible on any crisis-relevant timeline. The global biotechnology market is forecasted to reach more than $9 trillion by 2035.93 A recent CFR task force on U.S. economic security highlighted biotechnology as one of the most critical domains for U.S. economic and national security, warning that the United States is wielding last century’s tools in a twenty-first-century era of economic warfare.94
Structural Vulnerability
The risk of systemic competitive erosion across the entire value chain categorically distinguishes mAbs—and Archetype Two cases generally—from the upstream concentration risks of Archetype One. The risk of dependence, however, is the same. Over the past decade, China has deliberately built end-to-end biomanufacturing capacity, with WuXi Biologics as the primary vehicle. Its “Win the Molecule” model integrates services from discovery through commercial manufacturing, capturing U.S. client programs early and deepening dependence at every subsequent stage.95 A Biotechnology Innovation Organization survey found 79 percent of pharmaceutical companies use WuXi or other Chinese manufacturers, reflecting accumulated manufacturing know-how and client relationships that cannot be quickly unwound.96 WuXi alone added nearly 209 new integrated projects in 2025, roughly half from U.S. clients, and manages 74 late-stage and 25 commercial manufacturing projects.97 The scale and efficiency of WuXi’s model creates switching costs for firms that compound as development programs advance, and structural dependence that cannot be quickly reversed even when individual companies wish to do so.
The most alarming indicator rests in late-stage clinical data. At the Phase 3 level—the most direct leading indicator of immediate commercial competition—our analysis shows that Chinese firms hold roughly thirty-two of fifty-five mAb programs globally. The late-stage pipeline of this promising therapeutic class is increasingly a China story.
China biotech licensing activity surged to $137.7 billion in 2025, a tenfold increase from 2021.98 Biosimilars—similar, FDA-approved versions of existing biologic drugs—are the near-term future of the mAb market. Chinese manufacturers hold no current U.S. market share but have already obtained FDA approvals, positioning them for biosimilar entry as patents expire. Meanwhile, China has reduced its own upstream supply chain exposure by developing domestic alternatives to U.S. or European sources of single-use bioreactor bags, filters, and cell culture media. Meanwhile, U.S. and European dependence on Chinese contract research and manufacturing has only deepened and become more entrenched.
China could feasibly weaponize the emerging dependence in this product class to create economic, strategic, and political leverage. It could directly choke off supplies of an innovative medicine or countermeasure developed and manufactured in China on which U.S. patients have come to rely. The competitive loss of U.S. capacity to China at every stage of the value chain—discovery, development, manufacturing, and market access—also undermines U.S. economic security and capability to produce in a timely way the effective countermeasures and cancer medicines on which many Americans depend.
The U.S. policy response has been inadequate to this emerging dependence. The BIOSECURE Act, intended to address Chinese penetration of U.S. biopharmaceutical supply chains, was significantly diluted by the time it was enacted in December 2025. Even where legislative intent exists, commercial entities generate resistance sufficient to weaken policy before it takes effect.99
An Industrial Policy for Archetype Two Biologics: A Whole-of-Value-Chain Response
A credible Archetype Two industrial policy, which would address systemic competitive erosion across the full biologics value chain, needs to combine immediate interventions that reduce lock-in risk with future investments that rebuild U.S. and allied competitive capacity. All four pillars of the value chain should be addressed: R&D; clinical and regulatory science; manufacturing assets; and biologics workforce and data infrastructure. Any industrial policy should also ensure that secure U.S. and allied alternatives are commercially viable—not merely asking private firms to absorb concrete higher costs for abstract national-security benefits or compromising patient access.
The challenge is that China is building end-to-end biomanufacturing capability while U.S. market actors are optimizing for cost, speed, and convenience. Private life sciences firms and clinical trial sponsors have strong incentives to use established China-based or China-owned contract research, development, and manufacturing organizations (CRDMOs) for their integrated services, rapid execution, and lower costs. Market forces are more likely to deepen that dependence than correct it. The right time to address this emerging “rare earths”-type dependence in the U.S. pharmaceutical supply chain is now, before it fully arises.
The challenge of Archetype Two industrial policy is that China is building end-to-end biomanufacturing capability while U.S. market actors are optimizing for cost, speed, and convenience.
Near-Term Interventions
Make the BIOSECURE Act the floor, not the ceiling, in sustaining U.S. biotech supply. Implementation of the BIOSECURE Act is a necessary first step but does not fully resolve private-sector dependence, especially in biologics programs that are not yet federally procured but could become critical to U.S. public health, defense, or pandemic preparedness. Where national security is at stake—including medical countermeasures, pandemic-response platforms, and strategically important antibody or protein therapeutics—HHS, BARDA, DOD, NIH, and other funders should require China-risk plans across the board. Those plans should mandate qualification of an alternate U.S. or allied CRDMO before a predetermined development milestone. Contractual terms should also preserve technology-transfer rights, escrow or portability of process data, and contingency plans for relocating clinical or commercial production before pivotal trials or launch. This approach reduces lock-in with Chinese service providers without the potential chaos from forcing a sudden rupture.
Pilot a U.S. first-in-human trial acceleration model. The United States should create a first-in-human biologics acceleration pilot at the FDA and invest in clinical trial capacity at designated university medical centers and academic health systems of excellence, targeting shorter trial start-up timelines while maintaining strict scientific, ethical, and safety governance.
The FDA’s pre-investigational new drug (IND) application and processes create significant uncertainty for U.S. trial sponsors, leading them to over-engineer study designs to avoid the risk of clinical holds after IND submission and delays. Each trial site could have its own institutional review board (IRB) that reviews the clinical trial application and any subsequent substantial amendments. Approximately 2,300 U.S.-based IRBs currently exist. The combination of regulatory uncertainty and cumbersome processes make U.S. trials slower to initiate and more time-consuming to conduct, leading sponsors to look abroad for these critical FIH trials that establish potential commercial viability and are a crucial funding milestone for many early biotech ventures.
Australia and Norway each offer potential models. Australia accelerates first-in-human and early-stage trials through a risk-based clinical trial notification (CTN) and clinical trial application pathway, with the latter reserved for high-risk, novel therapies. Australia also offers a national single-application and tracking infrastructure (National One Stop Shop) as well as standardized operating procedures—streamlining timelines while preserving governance through mandatory IRB approval and drug regulatory inspection and backstop powers. Norway’s NorTrials program serves as a public-private national entry point linking industry sponsors with hospitals, feasibility support, central ethics committees, and funded clinical trial centers.
A U.S. pilot should adopt operational features of these two models without undermining patient safety or the scientific integrity of the trial. Congress and the FDA should also consider authorizing a narrow, optional CTN-like pathway for well-characterized drug platforms or lower-incremental-risk early studies, coupled with the FDA’s call-in authority, suspension authority, and strict eligibility criteria. A NorTrial-like approach to investing in U.S. university and hospital trial sites could boost capacity while ensuring the distribution of those sites supports the scientific objectives of a diverse trial subject population that can draw from rural areas as well.
Incentivize adoption of advanced biologics manufacturing technologies. The foundation of the U.S. system of biomedical innovation has been its partnerships between U.S. universities, entrepreneurs, and large drug firms, as well as its openness to tapping the best talent from abroad.100 That system is under siege and must be preserved and bolstered. Reducing the regulatory and commercial uncertainty that firms face when switching from legacy processes to adopt new technologies can help. The FDA should reduce this friction around adopting new technology by providing model comparability protocols for assessing new technologies and expedited review of post-approval manufacturing changes. The FDA’s existing Advanced Manufacturing Technologies Designation Program recognizes but does not fully resolve this inertia. On the demand side for a limited period, Medicare could pay a modest additional amount for qualifying biologics that meet defined manufacturing-resilience criteria. This could create incentives for manufacturers to accelerate the adoption of new technologies such as continuous manufacturing.
For biologic assets with national-security relevance, the FDA, BARDA, DOD, ASPR, and NIH should require trial sponsors and manufacturers to disclose critical supplier risk beyond the first tier. Covered inputs should include cell banks, plasmids, vectors, media, resins, filters, single-use bags and tubing, vials, stoppers, cold-chain inputs, analytical reagents, and quality-control assays. Disclosure requirements should focus on single-source dependencies, suppliers in foreign countries of concern, and inputs whose loss would materially delay clinical development, scale-up, or commercial supply.
Long-Term Interventions
Build allied-country CRDMO alternatives. The Development Finance Corporation should finance a portfolio of trusted CRDMOs in allied and partner countries. Though these would not immediately replicate the scale or integration of the largest China-based providers, they could create a healthier, more competitive market for biologics development and manufacturing services. DFC loans should target infrastructure expansion in allied country CRDMO networks where China risks are the highest.
Fund and authorize a National Biologics Manufacturing Excellence Center. The United States should establish a National Biologics Manufacturing Excellence Center to build the workforce and technical capabilities needed for advanced biologics production. Working with universities, community colleges, trade colleges, the National Institute for Innovation in Manufacturing Biopharmaceuticals (NIIMBL), and industry, the center should link classroom training, hands-on pilot-scale facilities, apprenticeships, and employer-recognized credentials to close existing U.S. workforce gaps.
Build AI-ready biodata and digital CMC infrastructure. Future competitiveness in biologics will depend heavily on data and artificial intelligence (AI) readiness. Building on the U.S. Data for the Bioeconomy Initiative, the White House should create an interagency effort, coordinated by the Office of Science and Technology Policy (OSTP) but involving HHS, FDA, NIH, National Institute of Standards and Technology (NIST), and BARDA, to develop a secure, AI-ready biodata and digital chemistry, manufacturing, and controls (CMC) initiative for comprehensively developing, producing, and testing biologics.101 The initiative should standardize data provenance, metadata, batch records, analytical methods, process-development data, and comparability datasets so that advanced analytics and AI tools can accelerate process development, technology transfer, regulatory review, and manufacturing troubleshooting.
Archetype Three: Dependence on China for the R&D Infrastructure for Sensitive Biotechnology
The defining characteristic of Archetype Three is vulnerability not in manufacturing fragility for existing drugs but losing control over R&D infrastructure underlying future pharmaceutical innovation, compounded by biosecurity risks accelerating with AI-driven sequence generation. Policy responses need to focus on biosecurity screening standards, intellectual property (IP) protection, and investment to create parity for U.S. synthetic biology platforms.
Case Study: Synthetic DNA
Synthetic DNA occupies a categorically different position from the other five case studies in this report: the vulnerability is centered on ongoing control over the R&D infrastructure that will underlie future pharmaceutical innovation on synthetic DNA, combined with biosecurity and IP risks that will grow more acute as AI-driven sequence generation accelerates.
Unlike prior case studies focused on supply disruption of existing products, the risks here are primarily forward-looking: control over synthetic DNA inputs and process is inseparable from control over the trajectory of U.S. biotechnology broadly. The Australian Strategic Policy Institute has a Critical Technology Tracker stretching back two decades that assesses “technology monopoly risk” as measured by concentration of scientific and technological research expertise within a single country. Synthetic DNA received the highest rating for risk of monopolization by China, just ahead of novel antivirals and antibiotics.102
Why This Product Is a National Security Concern
Synthetic biology—which leverages synthetic DNA sequences composed of individual nucleotides, the chemical building blocks of DNA—entails scientists designing, building, and testing synthetic DNA sequences in fields as varied and important as agriculture, industrial manufacturing, and therapeutics. DNA synthesizers allow researchers to make everything from heat-resistant crops to vaccines at the cost of just tens of thousands of dollars. The global synthetic biology market is projected to grow from $18.9 billion in 2025 to nearly $70 billion by 2033.103 As synthetic biology grows more sophisticated and scientists discover new ways to leverage the technology, control over the inputs and process becomes more important, especially as AI becomes capable of designing novel protein sequences and therapeutics.
Dependence on adversary-controlled sources of synthetic DNA creates exposure to intellectual property theft, data exploitation, and potential erosion of scientific dominance. China has already demonstrated an interest in accumulating genetic data—including through widespread COVID-19 testing, acquisition of U.S. genomic firms, and intra-PRC surveillance—suggesting its synthetic DNA industry is a potential intelligence vector.
Further, the programmable nature of synthetic DNA also makes it a potential tool of warfare: AI-enabled, mail-order synthetic DNA theoretically allows adversaries and bad actors to design, print, and build biological and chemical weapons with minimal effort.
Structural Vulnerability
Three firms—GenScript, Twist, and Integrated DNA Technologies—account for approximately 86 percent of global synthetic DNA supply, with WuXi as a major additional player. One of these firms—GenScript—is a Chinese company, while the other two have a significant presence in China.104 In addition, the nucleotide inputs for synthetic DNA production, both in house and at scale, appear to be highly concentrated in China, although exact data on the scope of the problem is not available.
As U.S. researchers increasingly route research and manufacturing orders through Chinese CRDMO platforms, those firms accumulate expertise, process knowledge, and build client relationships that deepen dependence over time—directly analogous to the offshoring dynamic that hollowed out small-molecule API manufacturing. Gradually, this dynamic helps raise Chinese biotech prospects at the expense of U.S. intellectual property and biotech research, as well as the nation’s development and manufacturing capabilities. Next-generation enzymatic synthesis technologies from U.S. and European firms could eventually diversify supply but remain pre-commercial and offer no near-term relief.
Speed and IP protection drive synthetic DNA procurement decisions—as synthesis fidelity has improved, quality is relatively standard across commercial synthetic DNA providers, removing a competitive lever that might favor higher-standard U.S., Japanese, or European producers. The risks most consequential for national security—biosecurity exposure, IP theft, genomic data accumulation—are diffuse, abiding, and national in character, borne by the public rather than the purchasers whose sourcing decisions create the vulnerability. No buyer has a commercial incentive to pay premiums for stronger screening standards.
Menu of Industrial Policy Tools: Archetype Three
Archetype Three reflects a different type of vulnerability than the upstream concentration and value chain erosion seen in the previous archetypes. The risk is that China will rapidly acquire control over the synthetic biology R&D infrastructure itself. Nucleic acid synthesis is a critical control point in modern biotechnology, and federal policy has already recognized screening as a key mechanism for reducing the risks of bioweapons.105
Therefore, the objective of U.S. industrial policy should be to prevent synthetic DNA and broader synthetic biology infrastructure from following the same offshoring and concentration trajectory that eroded U.S. API manufacturing and increasingly threatens biologics. The risks in this case, however, extend beyond supply disruption and include genomic data concentration, IP theft, and potential biosecurity threats.
Effective policy responses should operate across three layers: transparency and screening of new uses and technologies to identify potential risks and prevent misuse; investment in viable domestic and allied alternatives; and demand-side coordination to ensure the commercial viability of secure and new technology-based sources.
Effective policy responses should operate across three layers: transparency and screening of new uses and technologies to identify potential risks and prevent misuse; investment in viable domestic and allied alternatives; and demand-side coordination to ensure the commercial viability of secure and new technology-based sources.
Near-Term Interventions
Mandate DNA supply chain transparency and provenance disclosure. U.S. federal authority to oversee and set policy for DNA synthesis security is currently scattered across multiple agencies and offices, including the Office of Science and Technology Policy (which issued voluntary screening guidelines), the CDC (oversight of select agents), HHS and ASPR (which oversee preparedness), and the FBI. We agree with the National Security Commission on Emerging Biotechnology that the natural home for this activity is the U.S. Department of Commerce (DOC).106 The DOC has already exercised partial jurisdiction over the DNA synthesis sector under the Export Control Reform Act of 2018 and the Export Administration Regulations.
The federal government lacks visibility into DNA synthesis supply chains at the commercial level. The Office of Science and Technology Policy’s 2024 nucleic acid screening framework applied solely to federally funded research, leaving the broader commercial market unregulated.107 By executive order, commercial DNA synthesis providers operating in the U.S. market should be required to disclose corporate ownership structures, nucleotide input sourcing by country of origin, and order-routing practices. This disclosure should indicate whether orders placed with U.S.-branded entities are fulfilled through facilities in foreign countries of concern such as GenScript’s Singapore operations. The NIH, BARDA, and FDA should separately require that federally supported drug development programs disclose the source, ownership, and input provenance of synthetic DNA used in research, process development, and manufacturing. This parallels the Tier-N supplier risk disclosure recommended for biologics but applies to the R&D layer rather than the commercial manufacturing layer. Gradually, this data may enable FDA and Commerce to identify where synthetic biology input concentration is formed before it reaches the API-level dependence that characterizes Archetype One.
Mandatory biosecurity screening—performing sequence-of-concern checks against known pathogen and toxin databases—should be extended beyond federally funded research to all commercial DNA synthesis orders. If the Biosecurity Modernization and Innovation Act of 2025 were enacted, it would provide the legislative framework for doing so. Meanwhile, BARDA and NIH should condition procurement and grant funding on demonstrated compliance with screening standards, creating a de facto requirement for federally adjacent research. These screening standards should apply to where a DNA sequencing order is fulfilled, not just where it is placed. This requirement would serve to prevent third-country transshipment from operating as a compliance workaround.
Evaluate export controls on advanced DNA synthesis technology. The DOC Bureau of Industry and Security should evaluate controls on advanced DNA synthesis equipment and enzymatic synthesis platform technology to prevent transfer of next-generation capabilities to Chinese firms before domestic and allied alternatives are commercially established.108 Controls in this domain should therefore be paired with affirmative investment in U.S. and allied next-generation platforms (see Medium-Term Interventions), so that the window created by controls is used to build competitive alternatives rather than merely delay Chinese capability development.
Medium-Term Interventions
Invest in next-generation enzymatic DNA synthesis platforms. Current commercial synthetic DNA production relies mainly on phosphonamidite chemistry—a decades-old process that is cost-competitive at scale but depends on upstream nucleotide inputs heavily concentrated in China. Enzymatic DNA synthesis offers an alternative pathway operating under aqueous conditions, eliminating toxic chemicals byproducts, and enabling longer, more accurate sequences. But it remains pre-commercial and hasn’t yet reached the investment needed for broad adoption. ARPA-H and BARDA should fund a dedicated scale-up program for enzymatic synthesis platforms at U.S. and allied firms, structured as milestone-based contracts rather than open-ended grants, to accelerate the transition from laboratory demonstration to commercial viability.
Establish federal procurement preferences and offtake commitments for domestic and allied suppliers. Domestic and allied DNA synthesis capacity cannot be sustained on market economics alone if Chinese-linked competitors continue to undercut on price. BARDA, NIH, DOD, and ASPR should establish explicit procurement preferences and, where appropriate, advance purchase commitments or offtake agreements for synthetic DNA sourced from domestic or allied providers meeting defined biosecurity and provenance standards. NATO, which has a strategy for fostering responsible development of emerging biotechnologies and an innovation fund, could be a platform for these joint purchasing arrangements for defense and preparedness applications.109 Such joint allied commitments would signal durable demand to potential investors in next-generation platforms and prevent the market from consolidating entirely around the lowest-cost producers. Loan guarantees through the DFC or a domestic financing mechanism for capital-intensive platform investment by U.S. or allied firms can further help private firms raise capital for expanding DNA synthesis capacity.
Long-Term Interventions
Coordinate with allies on biosecurity standards and genomic data reference infrastructure.The United States should pursue harmonized biosecurity screening standards with key partners—including Australia, Canada, the European Union, Japan, and the United Kingdom—through existing frameworks such as the Biopharmaceutical Alliance, NATO, and emerging science-and-technology agreements. Harmonization should cover sequence-of-concern databases, screening methodology requirements, and provenance documentation standards, so that third-country routing cannot be used to circumvent requirements in any single jurisdiction. Coordination should also include reference biological material and “omics” data standards, thus ensuring genomics and other high-throughput biological data are interoperable and reproducible, reducing reliance on Chinese-linked platforms for foundational research infrastructure.
Cross-Cutting Requirements for Reducing U.S. Supply Chain Dependence
Governance Continuity
The dependencies outlined in this report cemented over decades across multiple administrations because no single institution had both the mandate and authority to act. SAPIR, Section 232 trade investigations, and the Biopharmaceutical Alliance are not only reactive measures and siloed approaches, but they rest on reversible executive action or discretionary appropriations. The 2008 heparin contamination crisis resulted in FDA regulatory arrangements, including a memorandum of understanding with China’s FDA, which failed to address the underlying supply chain concentration.110 The COVID-era DPA invocations for medical supplies were not followed up with durable manufacturing investment.
Meanwhile, the experience of the CHIPS Act is instructive, converting executive intent into statutory support and dedicated funding. The large bipartisan funding commitment was paired with a White House–level mandate, a dedicated implementation office, and stable multiyear appropriations that attracted talent, created institutional momentum, and established reporting requirements that made both progress and failure visible, and forced cross-agency alignment.111
The vulnerability of the U.S. pharmaceutical supply chain spans FDA, DOD, HHS and ASPR, the Department of Commerce, and the Office of the U.S. Trade Representative, without a single cabinet agency fully owning it. Reform of the U.S. pharmaceutical supply chain has diffuse stakeholders—patients, hospitals, GPOs, generic drug importers, pharmaceutical benefit managers—with sometimes competing agendas. Effective supply chain coordination is essential for items with national security implications as coordination failures between firms and fragmented government agency oversight lets dependencies and vulnerabilities go unaddressed. A dedicated coordinating body should sit at the White House level comparable to the National Security Council, Domestic Policy Council (DPC), or the National Economic Council to mandate action across agencies. It must be empowered to solve problems, not merely coordinate: directing agency timelines, fast-tracking regulatory approvals for new KSM and API suppliers, triggering emergency import pathways, overseeing the criteria for SAPIR procurement, and coordinating allied frameworks.
A U.S. Infrastructure for Measurement and Early Warning
These documented dependencies were visible in trade data and investigational new drug (IND) or abbreviated new drug application (ANDA) submissions for years before they reached crisis level. The United States simply lacked an institution with the mandate to look, the data infrastructure to see, and the authority to act on what it found.112 Without a strong monitoring function, even adoption of archetype policy tools recommended in this report will fall short—reacting to today’s vulnerabilities while allowing new ones to form undetected.
The barrier is organizational, not informational. The FDA should systematize IND, ANDA, and Drug Master File submission data that already exists on API and KSM manufacturing sites into a searchable database, accessible across coordinated agencies. Its mandate should include publication of an annual pharmaceutical competitiveness index tracking U.S., allied, and Chinese capabilities tier by tier. That would serve as an early-warning function that, had it existed, would have been capable of flagging forming dependencies in real time.
A monitoring function—similar to the publication of a drug shortage list but operating one to two tiers upstream—should flag markets at risk of developing vulnerabilities before they occur. Where concentration exceeds defined thresholds, FDA and the U.S. Department of Commerce should be required to notify the national security apparatus and initiate a remediation timeline.
Finally, the bilateral trade data comparison methodology utilized in this report (surfacing transshipment by comparing nominal and actual country of origin) should be institutionalized as a regulatory tool by FDA and DOC, rather than leaving it to academic or policy publications.
Allied Coordination as Force Multiplier
The Biopharmaceutical Alliance and the EU Critical Medicines Act represent meaningful commitments on paper, but without shared inventories, crisis allocation rules, mutual inspection recognition, and joint purchase commitments, allied coordination functions as a consultative forum, not a supply guarantee. Operationalizing these frameworks will make every archetype intervention more effective and cheaper: shared reserves reduce the required scale of domestic strategic stockpile holdings; mutual inspection recognition lowers regulatory costs and compresses timelines that currently make supplier diversification impractical during shortage conditions; and joint purchase commitments create demand signals that justify KSM-level investment in partner countries where commercial incentives are insufficient.113 DFC financing for allied CRDMO capacity applies with equal force to fermentation-based antibiotic inputs and porcine mucosa collection.
The main caveat is that all such mapping needs to include the KSM tier. If all the nations participating in the BIO-5—the biopharma partnership between United States, the European Union, India, Japan, and the Republic of Korea launched by the Biden White House—sourced identical intermediates from identical Indian manufacturers dependent on Chinese inputs, that coordination would only produce the semblance of diversification. Regulatory synchronization—expedited U.S. market-entry pathways for allied suppliers with equivalent GMP standards—stands as an immediate deliverable that could quickly make allied supply a credible buffer without any required legislation.
Security for the R&D Pipeline
This report’s concern over pharmaceutical manufacturing has now shifted a stage earlier to the innovation pipeline, establishing additional risk before structural concentration has even begun. The COINS Act establishes Treasury Department oversight of outbound U.S. investment in sensitive technology sectors but does not clearly touch on pharmaceutical and biotechnology licensing, the channel where dependence of the R&D pipeline is currently deepening.114 Every licensing agreement transfers not only commercial rights but process knowledge, clinical data, and development expertise, the same intangible attributes that, in manufacturing, accumulated in Chinese CRDMOs. Once embedded in Chinese agencies, they are not recoverable through downstream manufacturing policy.
Extending the COINS Act to cover national security–relevant licensing categories would apply the same logic used throughout this report but at a stage earlier. U.S. companies licensing Chinese assets in covered categories should retain technology-transfer rights, maintain a qualified domestic or allied development pathway, and disclose the arrangement to relevant federal agencies—a requirement that the archetype policy menus recommend for manufacturing.115
Restricting Chinese licensing without addressing U.S. research funding cuts, however, simply trades one vulnerability for another. Proposed NIH budget reductions, FDA staffing cuts, and scaled back investment in U.S. countermeasures are narrowing the U.S. R&D base while China expands its footprint.116 Rebuilding this domestic R&D infrastructure is the demand-side complement to any licensing restriction on China and a necessary condition under which allied and domestic alternatives become viable.
Pairing Innovation Funding With an Adoption Strategy
The United States has world-class research funding and a strong public-private innovation ecosystem—NIH, BARDA, DARPA, and ARPA-H, combined with private venture capital—that represent a genuine strategic advantage. The central policy challenge is converting those strengths into approved, scaled, and economically viable domestic and allied manufacturing capacity.
Newer platforms—continuous manufacturing, AI-enabled process optimization, advanced bioprocess analytics, cell-free and enzymatic synthesis, and modular biomanufacturing—can shift the cost and speed assumptions that have so far favored production in China and India, but U.S. manufacturers have weak incentives to adopt them once a biologic is approved because process changes require compatibility studies, new regulatory submissions, and risk of supply disruption.117
Technology funding policy should therefore be paired with adoption policy. The FDA and HHS need to reduce the friction of switching to advanced manufacturing through clearer comparability pathways, pre-negotiated post-approval change protocols, regulatory sandboxes for new modalities and precursors, and procurement preferences for domestically produced goods using advanced domestic technologies. AI compounds both the opportunity and the urgency: as it lowers the time and cost of biologics design, the bottleneck shifts toward who controls data, DNA synthesis, validation tools, and scale-up infrastructure. The fundamental issue is that R&D supply chain security is as central to biosecurity and competitiveness as commercial manufacturing itself.
Anticipating and Mitigating Unintended Consequences
The main risk with any industrial policy tool is creating new problems while trying to solve the original supply chain problem. A policy instrument that looks attractive as industrial strategy can raise procurement costs, create retaliatory procurement barriers abroad, lock-in obsolete capacity, or slow speed of innovation. Those unintended consequences should be anticipated and mitigated in policy development and implementation stages.
Buy-local provisions raise costs, which matter most in high-volume, price-sensitive generic markets such as statins, analgesics, and antibiotics. Local-content rules can also risk triggering reciprocal policies abroad. China’s volume-based procurement (VBP) illustrates how procurement mechanics can functionally exclude foreign firms even without explicit buy-local rules: in one VBP round, Pfizer, AstraZeneca, Roche, Gilead, Novartis, and other non-Chinese firms received zero volume.118
Volume guarantees also create overcapacity and lock-in risks if health-care demand shifts. COVID-19 vaccine advance purchase agreements scaled production rapidly, but post-pandemic demand reversals led to canceled or reprogrammed contracts in both the United States and the EU. The same risk applies to antibiotics, APIs, or biologics. The same lock-in risks extend to supplier networks, fill-finish relationships, and regulatory filings built around a specific platform.
Priority review vouchers (PRVs) and extended exclusivity avoid the need for direct appropriations but decouple rewards from policy goals. The transferability of PRVs allows them to be converted into tradable financial assets, which can be a significant incentive. Ipsen sold a PRV for $158 million in 2024, and Rocket Pharmaceuticals sold one for $180 million in 2026.119 Yet, PRVs are triggered by regulatory approval, not by sustained U.S. manufacturing, supply continuity, or follow-on R&D, which limits their effectiveness. Extended exclusivity carries similar downsides: a JAMA Health Forum analysis of four top-selling drugs found delayed generic entry led to approximately $3.5 billion in excess spending over two years.120
Stricter quality enforcement is most likely to prove the cleanest long-term tool. The FDA announced in 2025 it would expand unannounced inspections at foreign facilities producing essential medicines, building on pilots in India and China.121 The primary immediate risk is supply disruption if many foreign facilities simultaneously fail inspections.
Rejecting foreign clinical trial data can guard against approvals based on evidence of questionable quality, but a broad rule raises R&D costs and slows medical technology development. The sintilimab (an mAb used for various cancers) case is illustrative: an FDA advisory committee voted fourteen to one that additional trials were needed to demonstrate applicability to U.S. patients before approving the China-developed PD-1 inhibitor. The FDA subsequently issued a complete response letter to Lilly and Innovent, notifying them that their marketing application could not be approved in its current form.122 The better approach, however, could be clearer expectations for multiregional trials, bridging data, and U.S.-relevant comparators rather than blanket rejection.
Export controls on pharma R&D tools could protect choke points but can accelerate China’s domestic substitution. The Bureau of Industry and Security’s 2025 biotechnology rule restricting certain flow cytometers and proteomics mass spectrometers prompted China to develop domestic alternatives.123 The Illumina case is illustrative: after China barred Illumina from exporting sequencing instruments, the domestic Chinese sequencer market share rose substantially while Illumina’s fell.124 Export controls may buy time, but they can also accelerate the creation of competitive Chinese alternatives.
Conclusion
China’s dominance across critical U.S. pharmaceutical supply chains now extends well beyond legacy APIs and KSMs. Its position in monoclonal antibodies, biologics manufacturing, and synthetic DNA infrastructure has expanded rapidly, with full value-chain erosion accelerating in certain areas. Those vulnerabilities are not the result of normal market dynamics or an excessive U.S. focus on cost efficiency but rather decades of sustained and coordinated Chinese state investment, industrial policy, and strategic subsidization. Critically, Beijing has continuously demonstrated that it will weaponize supply chain concentration for foreign policy purposes, doing so through informal means that maintain plausible deniability and avoid the legal consequences of a declared embargo. The U.S. pharmaceutical chain is not exempt. It is increasingly the next frontier.
This report’s central finding is that the true scope of U.S. vulnerability is systematically underestimated and often poorly understood. Efforts to reshore or diversify U.S. supplies at the API or FDF stage only mask the near-total concentration upstream of key starting materials. Third-country transshipment obscures the degree of U.S. reliance on China for essential medicines, falsely suggesting that U.S. supply chains are resilient. Shortages of amoxicillin, heparin, and norepinephrine are already triggered by ordinary demand surges or disease outbreaks, without any adversarial action. These everyday shortages have already forced U.S. hospitals to ration care, delay surgeries, and substitute less effective treatments, leading to unnecessary patient deaths. These are not edge cases, but stress tests—and the United States has failed them. An effort by China to weaponize U.S. pharmaceutical dependence could do far worse.
The U.S. dependence on China for essential medicines has also moved beyond manufacturing into the innovation pipeline itself. Every licensing agreement signed with a Chinese biotech firm transfers commercial rights, process knowledge, clinical data, and development expertise that cannot be recovered through downstream U.S. manufacturing policy alone. The same dynamic that hollowed out small-molecule API manufacturing is now playing out in biologics and synthetic biology, but it is taking place at an earlier stage where intervention would be more effective and is more urgent.
For its economic and national security, the United States must respond with a package of coordinated measures built around the three archetypes of supply chain dependence.
Each of these industrial policy tools carries real risks: higher procurement costs, reciprocal retaliation, lock-in of obsolete capacity, or acceleration of Chinese domestic substitution. Disciplined, targeted implementation—calibrated to the specific archetype and its root cause—will be needed to mitigate these unintended consequences.
The tools for success exist, but it is past time to recognize that U.S. pharmaceutical supply chain security and resilience is a national and economic security problem that demands the same urgency and level of resources that the United States is devoting to reducing dependence on China for critical minerals and rare earths. The question now is whether the United States will manage to do so before a crisis makes the cost of decades of inaction all too visible to the American public and exceedingly painful for everyone touched by the health care system to bear.
Appendix: Summary Table of Recommendations
Advisory Committee
U.S. Pharmaceutical Supply Chain Study Group
Thomas J. Bollyky
Council on Foreign Relations
Luciana Borio
Council on Foreign Relations
Charles L. Cooney
Massachusetts Institute of Technology
Arnab Datta
Employ America
Jesse Dill
Capybara Therapeutics
Joel Dodge
Vanderbilt University
Rush Doshi
Council on Foreign Relations
Paul Friedrichs
RAND Corporation
Yanzhong Huang
Council on Foreign Relations
Olivia Kosloff
American Economic Liberties Project
Michael Kuiken
Stanford University
Kelvin H. Lee
University of Delaware
Timothy W. Manning
Georgetown University
Monique K. Mansoura
Beacon Biostrategies
Jude I. Nwokike
United States Pharmacopeia
Michael R. Schmidt
Princeton University
Ganesh Sitaraman
Vanderbilt University
Mariana P. Socal
Johns Hopkins Bloomberg School of Public Health
Vic Suarez
The Council on Strategic Risks
Rajeev Venkayya
Coalition for Epidemic Preparedness Innovations
Prashant Yadav
Council on Foreign Relations
This report reflects the judgments and recommendations of the authors. It does not necessarily represent the views of members of the advisory committee, whose involvement should in no way be interpreted as an endorsement of the report by either themselves or the organizations with which they are affiliated.
Acknowledgments
We are grateful to the members of the advisory committee for their time and insight. Although the views expressed in this report are our own, their feedback on the early version of the report not only expanded our understanding of pharmaceutical supply chain resilience but also strengthened our policy recommendations. We thank CFR President Mike Froman for his strong support of this project and CFR Senior Vice President of Studies Shannon O’Neil and Associate Vice President of Studies Stuart Reid for their review and incisive comments. Our special thanks to CFR research associates Benjamin Holtzman and Aarya Vaidya for their exceptional research support and seamless administrative coordination, and to Patricia Dorff, Caitlin Moran, and Tom Redburn for their guidance and editorial contributions. We thank Bloomberg Philanthropies and the Hewlett Foundation for their generous financial support for this project.
About the Authors
Thomas J. Bollyky is the Bloomberg chair in global health and director of the Global Health program at CFR.
Rush Doshi is the C.V. Starr senior fellow for Asia studies and director of the China Strategy Initiative at CFR.
Olivia Kosloff is a senior fellow at the American Economic Liberties Project.
Prashant Yadav is a senior fellow for global health at CFR.
Elena Every is the research associate for the Global Health program at CFR.
t

 Report
Report Report
Report Report
Report Report
Report Report
Report